
A “much-tweeted” manuscript was recently published on Biorxiv, investigating the barcode mis-assignment issues affecting HiSeq 3000, 4000, and HiSeq X, reported in our last newsletter. Please see the manuscript here: Sinha et al. 2017 http://biorxiv.org/content/early/2017/04/09/125724 .
As mentioned previously in our newsletter, the problem potentially affects low genome coverage sequencing studies and especially studies looking for low abundance mutations.
The authors come to the conclusion that “free index primers” in combination with the Exclusion-Amplification clustering chemistry of these sequencers are causing the barcode switching problems. Please note that all sequencing libraries in the reported experiments were generated with the Nextera XT chemistry that adds the barcode indices by PCR.
I would like to add my thoughts on this topic and the manuscript:
- The Sinha et al. study did analyze data from single-cell RNA-seq studies. These single-cell RNA-seq library preps require a lengthy protocol (SMART-cDNA generation plus NexteraXT) that is not typical for common sequencing library preps.
- The difficulty of these library preps is most likely the reason that the authors seem to have sequenced comparatively low quality libraries. The Bioanalyzer (BA) trace included below (edited from the Sinha et al. manuscript), shows that their libraries did indeed contain large amounts of free primers. Further the average insert size was a lot longer than recommended for HiSeq 4000 sequencing (eyeballing their trace, they likely had a mean fragment length of 800 bp or longer — in contrast to the recommended maximum of 500 bp). It looks that at most 30% of the library molecules were of the recommended length. Both of these library quality problems can very well impact the artifacts they are reporting. This Stanford lab seems to work exclusively with Nextera style library preps – which often result in libraries with out-of-spec inert sizes.
- The authors show, via primer spike-in experiments, that the concentration of free primers is directly proportional to the frequency of the observable artifacts (thus, the artifacts should be avoidable).
- Their findings do certainly ask for an in-depth study of the problem and for caution for projects in progress. It should be studied if the problem exists when the longer adapter oligos are employed (Truseq-style sequences) for the more common library preps using ligations.
- In conclusion: The data presented by the Sinha et al. manuscript indicate to me, that working with clean and high quality libraries will drastically reduce the occurrence of the described artifacts (or even eliminate them?). The latter assumption remains to be verified, but their artifact rates are very likely an exception. To some degree the manuscript shows: Ugly things can happen when sequencing really ugly libraries. Hopefully the Weissman lab has identified the cause of the problem, allowing a meaningful prevention (magnetic bead cleanups, gel-based fragments size selection, Exonuclease1 digestions as suggested on SEQanswers , and the use of uniquely dual-indexed adapters).
Some notes to the practices in our own lab:
- Our lab does not use the Nextera XT chemistry and for most preps barcoded adapters are added by ligation instead of PCR (exceptions are small-RNA-seq library preps, WGBS library preps, and Tag-Seq library preps). Thus, these libraries see at least two rounds of magnetic bead cleanups after the oligo additions, in contrast to the single cleanup for the Nextera preps. As a consequence, the oligos are removed with high efficiency in our preps. Further, we have recently added gel-based sequencing library size selections (with the Pippin HT).
- Our sequencing libraries are only sequenced when Bioanalyzer traces show all libraries to be “clean” (free of any visible primers and free of primer-dimers).
–> Based on the recommendations in the Biorxiv manuscript we will switch to indexed PhiX internal standards, and will employ uniquely dual-indexed adapters for the study types mentioned above.
–> Since we have shown previously that PCR-free sequencing libraries often harbor hidden primer-dimers in libraries that appear to be clean on the Bionalyzer, we will discourage the multiplexed sequencing of such libraries and will suggest to at least add a single PCR-cycle.
We are happy to assist with library clean-ups and size-selections.
The sequencing library trace from the Sinha et al. paper (slightly edited):
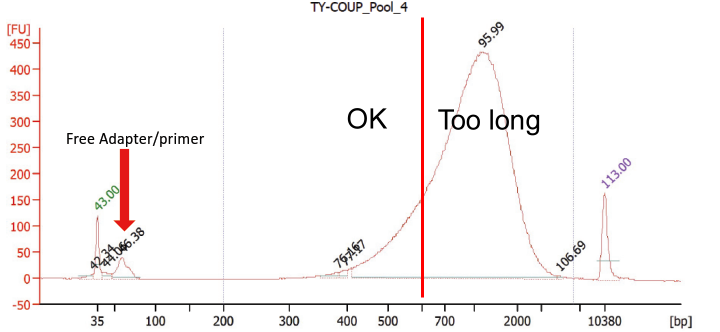
The figure shows a Bioanalyzer trace of the single-cell RNA-seq libraries studied by Sinha et al. The library contains a significant amount of free primers (arrow). In addition, the majority of the sequencing library fragments are significantly longer than recommended for HiSeq 4000 sequencing (all fragments to the right of the red bar).
EDIT (04-18-2017):
Illumina has just published a white paper on the issue: https://www.illumina.com/content/dam/illumina-marketing/documents/products/whitepapers/index-hopping-white-paper-770-2017-004.pdf?linkId=36607862
Please subscribe to our newsletter.