
3’Tag-Seq is a protocol to generate low-cost and low-noise gene expression profiling data. The protocol is also known as TagSeq, 3’Tag RNA-Seq, Digital RNA-seq, Quant-Seq (please note that most of these names have also been used for a variety of other protocols previously). In contrast to traditional RNA-Seq, which generates sequencing libraries from the whole transcripts, 3-Tag-Seq only generates a single initial library molecule per transcript, complementary to 3′-end sequences. For example for human samples, the restriction to a small part of the transcripts reduces the number of sequencing reads required by at least five times. In contrast to earlier “digital RNA-seq” protocols that were based on restriction digestions of cDNAs, the current protocol combines reverse transcription priming from the poly-A tail with random priming and adapter placement for the second-strand synthesis. In most cases up to 48 samples can be sequenced per HiSeq 4000 lane.
More than 90% of the RNA-seq studies carried out in our labs are analyzed exclusively for differential gene expression (DGE). The conventional full transcript RNA-seq protocols generate more data than needed for this specific purpose, but they also allow for splicing analyses. The complexity of the standard RNA-seq data is not an advantage if the aim of the project is only DGE analysis – 3’Tag-Seq might actually be the superior tool for this application (DGE). In our experience the 3’Tag-Seq data have so far shown exceptionally low noise as well as insensitivity to RNA sample quality variations.
This example MDS plot shows an analysis of 3’Tag-Seq data of macrophage cells exposed to three types of bacterial infections and mock-infections at two time points. The analysis distinguishes the responses to the individual bacterial species and the duration of the infections. Even the reactions to the mock-infections are clustered by time points.

We are currently offering 3’Tag-Seq as a low cost custom sequencing service but are planning to offer 3’Tag-Seq services soon at simple per-sample recharge rates — including both library preps and sequencing. In the long run the services can also include a basic differential-gene-expression analysis.
Advantages of 3’Tag-Seq:
- low noise gene expression profiling
- less sensitive to RNA sample quality/integrity variations (compared to poly-A enrichment protocols)
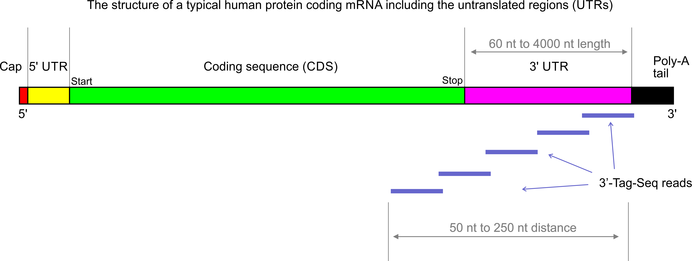
- >99% strand-specific; same direction as mRNA transcripts
- requires significantly lower numbers of sequencing reads
- single read sequencing is sufficient
- simpler library prep protocol
- costs about half or less compared to standard RNA-seq
- costs lower than, or comparable to, microarray analysis
- much higher dynamic range compared to microarrays
- we routinely sequence 48 libraries per HiSeq lane; for soBarclays
- for very low input or high depth sequencing of 3’Tag-Seq libraries UMI‘s (unique modular identifiers) can be incorporated
- Batch-Tag-Seq packages: simple pricing scheme and simplified planning of experiments
Disdavantages of 3’Tag-Seq:
- data analysis requires a reference genome with good annotation (including UTRs)
- only applicable to eukaryotic samples
- data do not contain any transcript-splicing information
- protocol is (a bit) more sensitive to chemical contaminants (spin column cleaned RNA samples are recommended)
For high-throughput 3’Tag-Seq library generation we require pure total RNA samples at a concentration of 100 ng/ul (best submit 10 ul at 100 ng/ul). For custom 3’-Tag-Seq library preps the input amounts can be a low as 10 ng total. The RNA samples for this protocol need to be isolated or cleaned-up by spin-column protocols. Please also see the sample requirements page.
3′-Tag-Seq libraries are sequenced by single-end sequencing on the HiSeq 4000 or the NextSeq.
Please note that 3’Tag-Seq libraries generate lower read numbers on the HiSeq 4000 (about 320 million reads per lane) compared to standard RNA-seq libraries. Since the DGE analysis of tag-Seq data requires much lower read numbers this is usually not a problem.
The libraries will be sequenced on Illumina HiSeq 4000 or NextSeq 500 sequencers with single-end 80 or 90 bp reads (SE80 or SE90). Please note that for some analysis pipelines it is recommended to trim off the first 12 bases from the reads. We will provide the full length data. Trimming is not necessary if you are using a local aligner (like STAR or BBmap). The sequences can be trimmed easily, for example with the “reformat” command from BBTools. In case UMIs are incorporated, the first 6 bases of the forward read represent the UMI, followed by a common linker with the sequence “TATA”, followed by the 12bp random priming sequence. It is recommended to transfer the UMI sequence information to the read header and trim the first 22 bases from each read with UMI-TOOLS or custom scripts. The same software can be used to remove PCR-duplicates after the alignments.
Please also the 3’Tag-Seq data analysis recommendations and this note on working with degraded RNA samples.
Comprehensive Batch-Tag-Seq info for download with UC pricing.
← FAQ