
As sequencing output increases and experimental scales are growing, generating libraries for sequencing is often the rate-limiting step. We are happy to discuss the options and protocols suitable for your specific research projects. We can prepare standard as well as specialized libraries of various types, including genomic DNA with different size inserts, RNA-seq with Ribo-depletion or strand specific options, exome capture, ChIP-seq, and microRNA-seq. We have streamlined and automated library preparation and can now generate up to 96 different barcoded libraries using the IntegenX Apollo 324 robot and Caliper Sciclone G3 for consistent quality and rapid turnaround. We can also provide training and access to the robots if you wish to use the instruments yourself for large-scale projects.
Starting material for Illumina library construction is usually double stranded (ds) DNA from any source: genomic DNA, BACs, PCR amplicons, ChIP samples, any type of RNA turned into ds cDNA (mRNA, normalized total RNA, smRNAs), etc. Pretty much anything you can think of that ends up as, or can be turned into, dsDNA. This dsDNA is then fragmented (if it is not already, as in ChIP). The average fragment length should not exceed 600 bp (HiSeq 2500, MiSeq) or 350 bp (HiSeq 3000). Then the ends are repaired and ‘A’ tailed, adapters are ligated on, size selection is carried out, then PCR performed to generate the final library ready for sequencing. Different library types might vary in the details (such as PCR-free library) but this is the basic workflow. An excellent forum for sequence related questions of all kinds on all platforms is the Seqanswers.com forum.
DNA/RNA sample quantification and purity
The input DNA and RNA quantities specified below and in this table apply if the samples are quantified by a fluorometric method (e.g. Qubit, PicoGreen, RiboGreen). Fluorometry provides advantages in precision and specificity (e.g. DNA dyes will not bind to/measure RNA). If a spectrophotometer (e.g. Nanodrop) is used, we suggest submitting twice the requested amount of sample since this type of measurement is often unreliable. In any case, sample amounts higher than the minimum requirements will improve the library complexity. Spectrophotometer readings are very useful to assess the purity of samples. For DNA samples the 260/280 ratio should be between 1.8 to 2.0 and the 260/230 ratio should be higher than 2.0. For RNA samples the 260/280 ratio should be between 1.8 and 2.1 and 260/230 ratio should be higher than 1.5. Values outside of these ranges indicate contamination. The Real-time PCR Core can carry out DNA as well as RNA extractions.
Sequencing Library Prep Services: Sample Requirements
Please also see the Comprehensive Requirements Table
DNA Based Libraries
Performance specifications of libraries we produce depends on the source material. Genomic DNA, double strand cDNA libraries, BACs, or other material available in microgram quantities will generate quality libraries nearly every time.
Guidelines for Submission of Library-Worthy DNA
Provide 2 ug or more of high quality DNA (concentration > 50 ng/ul, OD 260/280 close to 1.8; 260/230 ratio >2.0) in EB or TE buffer (EB buffer preferred), or molecular biology grade water. Library construction can also be attempted from less input material, with caveats. If the total input material for library prep is below 100 ng special library prep protocols need to used. For PCR-free libraries sample amounts of 5 ug DNA are recommended; working with less is possible.
ChIP Libraries
We offer library construction from chromatin immunoprecipitated material. For these more complex experiments, discussions with Core personnel regarding suitability of starting material and construction strategy are recommended. No guarantees are offered with this library service, other than we’ll do our best! For general background, the ChIP-Seq Data Technical Note and ChIP-Seq DataSheet from Illumina may be of interest.
Mate Pair Libraries
The sequencing of Mate Pair libraries generates long-insert paired-end reads. The libraries are generated by self-ligation of long DNA fragments and labeling of the junction sites to generate chimeric library molecules that bring together sequences that were originally 2kb to 12 kb apart. We are using the Illumina Nextera Mate Pair kit which employs a transposase enzyme to fragment as well as end-tag the DNA in a single step. The tags are biotinylated and thus allow for the selection of junction sites containing fragments. In contrast to older mate pair library protocols, the Nextera kit is very reliable with the exception of the sizing of the initial fragments. As with all other long DNA fragment analyses, the DNA quality matters. Please email us an gel-image before submitting the DNA samples. The samples should run as a band of 20kb size or longer on agarose gels.
The Nextera kit offers two protocols: the “gel-free” version (1 ug input), which is mostly of interest when only little input DNA is available. The sizes of the mates-pair fragments from this protocol usually range from 1.5 kb to 10kb. Surprisingly the SSPACE scaffolder can still work with these data.
The “gel-plus” version requires a minimum of 4 ug input DNA (and 4 times the reagents) and uses gel extractions to size select fragments within a range of +- 700 bp for shorter mates and within +- 2kb for longer mates of up to 10 to 12 kb. Due to the uncertainties of the fragmentation please submit at least twice the amount of sample.
In theory the fragment sizes resulting from the tagmentation are only dependent on the input DNA amount. In praxis the fragment lengths vary considerably between different DNA samples of similar amounts. This variability between samples can be observed even after precise DNA quantification by fluorometry. The reactions are tune-able for aliquots of the same sample, though. Especially if very specific size ranges are desired it is often necessary to repeat the tagmentation reaction with adjusted DNA amounts. We might then combine similar sized gel extraction fractions from two tagmentation reactions to generate libraries of high complexity for the desired size ranges. Please let us know how important specific insert size ranges are for your project.
Because of the difficulties to predict the fragment size ranges, we are quoting mate pair recharge rates including two tagmentation reactions. If we can generate the desired library with a single tagmentation, we charge the lower rate of the one-tagmentation library prep.
Target Enrichment
Numerous companies provide services and platforms that generate whole exome or target amplification. We offer the Fluidigm Access Array, which employs nanofluidics for cost-effective target selection to generate barcoded amplicon libraries that are ready for Illumina sequencing. Sequence Capture Libraries are those in which particular genomic regions are enriched after indexed library generation and sequencing. This strategy allows focused, very deep sequencing and can be implemented for a number of applications. Several companies offer platforms that can generate such material, including Illumina, RainDance, Agilent, NimbleGen, and Fluidigm. Technical information on the Agilent, Nimblegen, RainDance, and Qiagen (which uses a PCR based, not hybridization capture strategy, for enrichment) systems are available but not guaranteed to be up to date. Think of them as a starting point for further investigation (we have the contact info for company reps if needed) and solely informational (no implied endorsement etc.).
RNA-Seq Libraries
Something of a misnomer because all the libraries end up as DNA, but this refers to the starting material. We offer RNA-seq library preparation, with a number of options such as ribo-depletion, poly-A enrichment, strand-specific libraries as described below as well as micro-RNA (miRNA) and small RNA library preps.
Guidelines for Submission of Library-Worthy RNA
Provide at least 1 ug (2 – 5 ug preferred) of total RNA at a concentration of at least 50 ng/ul (1 ug for Poly-A enrichment; 2 ug for ribo-depletion libraries; using less starting material is possible, but we can’t guarantee results). Please make sure that your RNA isolation protocol employs a DNAse digestion step or other means to remove DNA from the sample. On an agarose gel, DNA contamination will be visible as a smear of band of fragments considerably larger than the RNA (>10 kb). To verify the purity of the RNA samples the 260/280 ratio should be between 1.8 and 2.1 and 260/230 ratio should be higher than 1.5. Poly-A enrichment, ribo-depletion and strand specific library prep are among the commonly requested types of service (more technical details on this appear below). We suggest following the recommendations from Illumina – for human samples use total RNA with a bioanalyzer RIN score of 8 or better, for plant material RIN numbers can be lower and tissue-specific (this is mainly a function of the chloroplast content). Libraries for slightly degraded RNA samples should be prepared using ribo-depletion protocols. If possible please avoid RNA extraction protocols involving Trizol or related phenol containing reagents (silica column based kits are less likely to retain contaminants). If using Trizol, protocols that contain a column based cleanup (e.g. Direct-zol, TRIzolPlus) have to be used. Please note that an additional column cleanup is mandatory for RNA samples isolated from PAXgene tubes or with PAXgene kits. RNA samples should be eluted in molecular biology grade water, always stored in a -80 degree freezer and shipped on dry ice. All RNA samples require a Bioanalyzer sample QC (or equivalent). Such QC traces can be submitted by the customers or we can run the QC for a fee instead.
Poly-A Enrichment
Total RNA samples can contain up to 90% ribosomal RNA sequences, which are uninformative for transcriptome or gene expression studies, while mRNAs typically make up only 1 to 2% of total RNA. Thus the enrichment of samples for mRNAs is highly desirable. Poly-A enrichment is the most commonly used method to enrich mRNA sequences from eukaryotic total RNA samples; mRNAs are selected by hybridization to poly-T oligos bound to magnetic beads.
Ribosomal RNA Depletion
There are multiple commercially available kits to remove ribosomal RNA from your total RNA. The main reason for rRNA depletion is to reduce highly abundant ribosomal RNA especially when transcripts do not carry polyA (bacterial RNA), and also when you desire to retain all long non-coding RNA (lncRNA) and polyA classes of RNA in your sample. Commercial kits containing rRNA removal solution are available for different types of total RNA; they include human, mouse, rat, bacteria (gram positive or negative), plant leaf, plant seed and root, and yeast. Ribo depletion protocols can further enable the analysis of slightly degraded RNA samples. We ask for at least 2 ug of total RNA for the preparation ribo-depleted libraries. As always libraries can be generated from less material, but the complexity can suffer.
Micro RNA and Small RNA Libraries
We offer library construction for micro and small RNAs from total RNA using the Illumina protocol and reagents. We size select the libraries with high precision using the Blue Pippin system. The minimum recommended amount of total RNA required for these preps is 1 microgram (recommendations for humans samples). Since the total RNA composition can vary widely between tissues and organisms, please aim to provide at least 2 ug of total RNA. Please also take care that you RNA isolation method actually retains micro and small RNAs. The total RNA samples should be submitted in molecular biology grade water at a concentration of 200 ng/ul. High quality RNA is recommended (the total RNA samples should have RIN scores of 8 or higher according to a Bioanalyzer QC) and should have been DNAse treated before sample submission.
Strand-Specific RNA Libraries
By default we generate strand-specific RNA-seq libraries in the Core. Please let us know if you would prefer the traditional non-stranded library prep instead. Strand-specific (also known as stranded or directional) RNA-seq libraries substantially enhance the value of an RNA-seq experiment. They add information on the originating strand and thus can precisely delineate the boundaries of transcripts in regions with genes on opposite strands, and can determine the transcribed strand of non-coding RNAs. During the cDNA synthesis dUTP is incorporated in the second-stand synthesis. After adapter ligation the dUTP-containing strand is selectively degraded, to preserve strand information for RNA-seq. The forward read of the resulting sequencing data thus represents the “anti-sense strand” and the reverse read the “sense strand” of the genes (for Trinity transcriptome assemblies the “–RF” orientation flag should be used).
Other Library Considerations
PCR-Free Libraries
Libraries generated without amplification will reduce library prep biases. Thus, they can improve the sequencing coverage of genomic areas such as GC-rich regions, promoters, and repeat regions, and enhancing the detection of sequence variants. Please note that PCR-free libraries are more difficult to QC and quantify (please see the bottom of the page) and that the yields tend to be lower for these libraries compared to amplified libraries (10-15%). PCR-free library prep will also require a greater amount of starting material (>5 fold).
Indexed Libraries
Indexing, also called barcoding, allows for the sequencing of multiple libraries in a single lane, i.e., multiplexing. By default all libraries generated by us have a barcode. Multiplexing is required when the typical lane output of 15-25 million reads from the MiSeq, 120-180 million reads from the HiSeq 2500, or 260-310 million reads from the HiSeq 3000 is greater than required for a single library (e.g., in sequencing BACs, PCR generated fragments, small microbial genomes, transcriptomes, exome, ChIP, and small RNA applications). Multiplexing is also the best way to minimize potential lane-to-lane sequencing variation, as all of your samples are subject to the same sequencing conditions. For example, if you require two sequencing lanes for six samples we recommend 6-plexing and sequencing over two lanes, instead of 3-plexing per lane. The principle is that short nucleotide “barcodes” are appended to each library using specific adapters containing those sequences. Libraries containing different indexed adapters are then constructed, quantified, pooled in equimolar amounts, and sequenced. Deconvoluting the barcodes informatically allows multiple libraries to be sequenced in a single lane at a potential cost and time saving. To date, two methods have been exploited for this: using the commercially available indexing kits (Illumina TruSeq, Nextera, or Bioo Scientific) or synthesizing your own adapter oligos with your own barcodes. With the Illumina TruSeq v2 Library Prep Kits A and B you can use up to 24 different barcodes per kit to multiplex up to 48 libraries. Bioo Scientific offers Illumina-compatible barcodes (NEXTflex) with up to 96 barcodes. The Nextera kit (Epicentre/Illumina) uses dual indexing and transposon mediated fragmentation (‘tagmentation’) followed by PCR amplification to integrate barcoded adapters (so a PCR-free library is not an option using the Nextera kit). The dual indexing/adapter tagging strategy (with up to 12 indices available for adapter 1 and up to eight indices for adapter 2) permits up to 96 unique dual index combinations.
Homemade indexing has been used successfully by multiple users. Please avoid the “in-line” barcoding strategy and use Truseq or Nextera-style adapter designs instead (i.e. the barcodes are read in a separate read and do not interfere with cluster registration). It is important to ensure that the base composition of the indices are balanced to optimize the ability of the image analysis software to distinguish signals.
Libraries: Make Your Own
Library construction involves DNA fragmentation (if necessary, depending on the nature of the initial sample), enzymatic treatment of the DNA to repair and A-tail the fragments, ligation of sequencing adapters to these fragments, then subsequent PCR amplification (or skip this for PCR-free libraries), with or without size selection depending on the application. See below for more information on these various aspects of library construction. We also offer Next-Gen Library Prep Training Workshop for comprehensive hands-on training on how to prepare high quality libraries for Illumina sequencing.
Fragmentation
DNA to be made into a sequencing library must first be converted into small fragments. The average insert length should not exceed 600 bp (HiSeq 2500, MiSeq) or 350 bp (HiSeq 3000). There are several methods for doing this, each with attendant pros and cons. Many protocols and centers rely on and recommend a fragmentation device from Covaris, which uses adaptive focused acoustics to break the DNA into appropriately sized fragments. The Covaris E220 can meet the demands of high throughput library production. We primarily use a Covaris E220 and Diagenode Bioruptor NGS (or Bioruptor UCD-200). Access to these instruments is available through the Core, with the usual training and sign up guidelines for Core-available equipment in effect.
Basic DNA and RNA Library Protocols
We use library kits from Illumina, Wafergen, Kapa Biosystems, Bioo Scientific, CloneTech, and NuGen as a source of the fragment repair, tailing, and amplification enzymes. There are a number of other next-gen related products currently being used in the research community. For mRNA-seq libraries we are currently using the stranded Illumina kit. New products are out there and we encourage you to do research if RNA-seq libraries are of interest. In particular, ribosomal RNA depletion protocols that integrate with Illumina kit, and novel RNA amplification and library production resources from NuGen and CloneTech, have expanded the services we offer and will no doubt continue to do so. In other words, keep checking this site to see how things evolve.
The Illumina Adapter Oligonucleotides
The oligonucleotide sequences of the Illumina adapters are available here. Illumina tends to sell their adapters only in conjunction with library prep kits. Other vendors of fully compatible ready-to-go adapters include Bioo Scientific. Custom synthesis from companies like MWG-Eurofins, Bioneer, and IDT is another valid option. Two things to note – the “top” Indexed adapter (starting with GATC) must be phosphorylated, and the “bottom” Universal adapter can be synthesized with a special linkage between the 3′ terminal T and the preceding C. This phosphorothioate linkage renders the overhanging T (after annealing the top and bottom adapter oligos) more nuclease resistant, diminishing the probability of adapter dimers (more on adapter dimers below).
Libraries: Quality Control (QC)
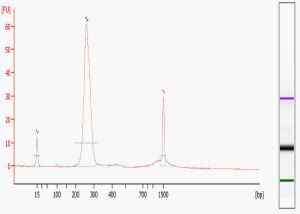 Library quality is the single most important determinant of the success of your sequencing run, both in terms of the number of reads generated (quantification) and the validity of the sequence obtained (content). Methods for construction and analysis continue to evolve; while now somewhat dated a useful early paper by Quail et al. from the Sanger Institute lists a number of improvements over the standard Illumina protocols in library preparation and analysis. If you construct your own libraries you may want to download this paper and a supplementary methods table for the many practical issues covered. We carry out two QC measures on all libraries sequenced in our Core – examination on the Agilent Bioanalyzer, and quantification using the Kapa Biosystems Illumina Library Quantification Kit.
Library quality is the single most important determinant of the success of your sequencing run, both in terms of the number of reads generated (quantification) and the validity of the sequence obtained (content). Methods for construction and analysis continue to evolve; while now somewhat dated a useful early paper by Quail et al. from the Sanger Institute lists a number of improvements over the standard Illumina protocols in library preparation and analysis. If you construct your own libraries you may want to download this paper and a supplementary methods table for the many practical issues covered. We carry out two QC measures on all libraries sequenced in our Core – examination on the Agilent Bioanalyzer, and quantification using the Kapa Biosystems Illumina Library Quantification Kit.
The Bioanalyzer provides a detailed visual examination of the libraries. The “perfect” library electropherogram, pictured here, shows a single peak of the expected molecular weight. Common additional forms include primer dimers (at around 80-85 bases), adapter dimers (around 120 bases), and broader bands of higher MW than the expected peak. Primer dimers, minimized by the use of magnetic beads, are not a problem unless they completely dominate the reaction. Adapter dimers can be a problem because they will sequence much more efficiently. As a result, whatever the proportion of adapter dimers in your library will be seen as an even higher percentage of reads in your final data files. Adapter dimers can be minimized by adjusting the adapter:insert ratio during library construction and exercising care in gel extraction or other size selection steps. The larger MW, typically more hump-shaped forms that are visualized on the Bioanalyzer are probably a result of excess amplification during the final PCR step. While some amount of these are tolerable, if they are too prominent then the library should be re-amplified from the gel extracted material.
We use a qPCR assay for library quantification – the Kapa Biosystems qPCR assay is run on all the libraries we sequence (and is included in the sequencing price). This has allowed us to provide much more consistent cluster values, which translates to more consistent read numbers. For long runs in particular it’s essential to maximize the data recovered given the time and money involved, which is why we recommend this quantification so strongly.
Library Requirements, Submission, and Storage
Submission
We must receive electronic (dnatech@ucdavis.edu) and print copies (submitted together with samples) of the appropriate submission form. All customer submitted libraries should be accompanied by Bioanalyzer (or similar) traces. If no traces are submitted we will carry out the Bioanalyzer analysis for a fee. Please visit the Sample Submission & Scheduling page to download submission forms and for more detailed instructions. The same form is currently used for both library preparation and library sequencing submissions. Please contact us if you have any questions about the required information; it is essential that you fill in all the information to minimize the chance for error on these expensive and time consuming experiments. One thing we need to know is the approximate insert size desired; in the absence of specific preferences we recommend about 220 bp for most mRNA and DNA libraries, but insert sizes should be larger for the longer read MiSeq runs. For certain applications, such as de novo assembly, a range of sizes may be desired and we can accommodate that. But again, we strongly recommend verifying the suitability of these values for the experiment you are trying to do.
Sequencing Library Requirements
The standard requirements for library submission are at least 15 ul volume at a concentration of 5 nM (e.g., 2.3 ng/ul given a 700 bp library). More volume and/or higher concentration is welcome. We can work with less library (down to 1 nM), but the quantification becomes less reproducible, the library becomes less stable, and relatively larger amounts of library DNA stick to the sides of the storage tube. Lower sequencing yield is the likely outcome for library concentrations 1 nM or less, and we cannot guarantee the data quantity or quality for such libraries. The best buffer to store and submit libraries is 10 mM Tris/0.01% Tween-20 ph=8.0 or 8.4, but EB buffer is also acceptable. If possible please use 0.6 ml or 1.5 ml low-bind tubes. If you do not provide a Bioanalyzer trace (or equivalent) of your library, we will do this for a fee. Please note, the DNA insert size(s) should not exceed 700 bp and most Illumina adapters add about 120 bases to the fragment length as observed on the Bioanalyzer. When submitting your libraries for sequencing, please use our Illumina Sequencing Submission Form (hard copy with samples, and email to dnatech@ucdavis.edu), provide the Bioanalyzer profile, library prep methods, and index sequences used. We will measure the quantity of your libraries using real-time PCR (included in the sequencing price).
Libraries for HiSeq 3000 sequencing – The latest generation of sequencers has more stringent library requirements and requires higher library concentrations. The average insert size should ideally be 350 bp and the “tail” of longer fragments should not exceed 550 bp. The new clustering chemistry is more sensitive to adapter dimers: a 5% adapter-dimer contamination can result in 60% of the reads coming from these dimers. Thus it is very important that there is no indication of an adapter-dimer peak (around 120 bp) on the Bioanalyzer trace. Our preferred requirements for library submission are for at least 15 ul volume of 5 nM concentration (e.g., 1.6 ng/ul given a 470 bp library). More volume and/or higher concentration is welcome. Lower sequencing yield is the likely outcome for library concentrations 2 nM or less, and we cannot guarantee the data quantity or quality for such libraries.
PCR-free Libraries QC – The quality of these libraries is difficult to assess. The adapters of these libraries are partly single-stranded. Thus they tend to migrate slower than the fully double-stranded amplified libraries on the Bioanalyzer. In most cases the libraries appear to be 70 to 100 nt longer than they actually are – however the bioanalyzer traces can also be off by far larger margins (e.g. 500 bases). To be sure about the actual library fragment lengths we highly recommend to PCR-amplify an aliquot (1 ul) of the libraries with 8 PCR cycles and run both the PCR-free and the amplified sample on the Bioanalyzer. If multiple PCR-free libraries will be pooled, you might consider quantifying the individual libraries by qPCR before pooling.
Custom Sequencing Primers Please note that these are used only for a small minority of sequencing projects. Custom sequencing primers need to be submitted at a concentration of 100 uM and a volume of 20 ul each together with the libraries. Please make sure that the sequencing primer design fits the chosen Illumina platform. Miseq and Hiseq platforms use different annealing temperatures.
Scheduling
Once your library, or library raw material, is ready, you should deliver it as soon as possible to get the next available slot in the queue. Runs occur as we fill up the two (rapid mode) or eight (high-output mode) lanes on a HiSeq flow cell, and the timing on this can vary depending on service type and Core activity. For MiSeq runs the turnaround time is typically five to eight days, while you should allow three to five weeks for HiSeq sequencing; in both cases allow an extra one to two weeks for library prep. The HiSeq sequencing scheduling calendar is now available on our website.
Sample/Library Storage Policy
Please let us know if you would like to pick up your samples/libraries after they have been sequenced and we will be happy to accommodate you; otherwise, due to space limitations, they will be stored for only six months after sequencing runs have been completed.