
Please see this page for updated information:
http://dnatech.genomecenter.ucdavis.edu/hiseq-3000-sequencing/
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
First Data > 300 million reads per lane
The first two runs of our Hiseq 3000 look very promising. The specs on Illumina’s website do suggest a yield of between 260 and 310 million reads per lane passing filter. Our first PE100 run including 5 PhiX lanes yielded between 318 and 387 million reads (a mean of 339 million clusters passing filter). The second run and third runs (PE150; all customer samples) averaged 343 million and 378 million clusters passing filter. The read/cluster numbers have basically doubled compared to the HiSeq 2500. The error rate was 0.35% and 0.61% for forward and reverse reads respectively for the PE100 run. The yields are higher than expected and the read qualities look good as expected.
Previously the “Patterned Flowcell/Exclusion Amplification” technologies, which allow the high yields, were only available on the X Ten sequencers for a single application, the re-sequencing of human genomic samples. The HiSeq 3000 will be used to sequence a much wider range of libraries types. It might require some adjustments to the library prep protocols to optimize them for the new generation of sequencers (e.g. size selection to remove very large fragments). We will assist you to make the libraries compatible.
In case you are interested in HiSeq 3000/HiSeq 4000 data we have uploaded a complete lane with PhiX data (including the reads not passing filter) here:
https://bioshare.bioinformatics.ucdavis.edu/bioshare/view/mq85kgzwb5cnl7k/ (WGET and web browser download options will work without setting up an account with Bioshare). Please note that for analysis purposes we have not filtered the data. Thus there are 482,680,800 reads per file. The sequencer generates “reads” for each single nanowell – no matter if it is loaded or not. Usually the data from the empty wells would be filtered out.
Please note that several run metrics are calculated differently for the HiSeq 3000/4000. The “Clusters Passing Filter” metric is now calculated as a percentage of the number of wells and thus is seemingly considerably lower; values between 55% and 70% are expected. Each flow cell lane has 482.68 million nanowells. Some of our customers were surprised by the demultiplexing metrics which are now delivered as “laneBarcode.html” files. The “Top Unknown Barcodes” table therein usually begins with more than 100 million barcode reads of “NNNNNN”. This is not a sign of any sequencing problems. This figure reflects merely the unused nanowells and more than 150 million unused nanowells are fully expected.
The quality scores are now binned to reduce the file sizes; the scale stays the same, though. (Added 07/2015) The quality scores now reach up to the code K (43) which is 1 score higher than was common previously. Some older bioinformatics tools might have problems with this score. In this case it is helpful to convert all “K”s in the quality score line with “J”s. This will not cause any loss of information. In unix/linux/mac environments you could use for example this command on the FASTQ files (thanks: Dylan Storey): sed -i '4~4 s/\([]{}\|~\\\^_`[K-Za-z]\)/J/
Some Q30 plots:
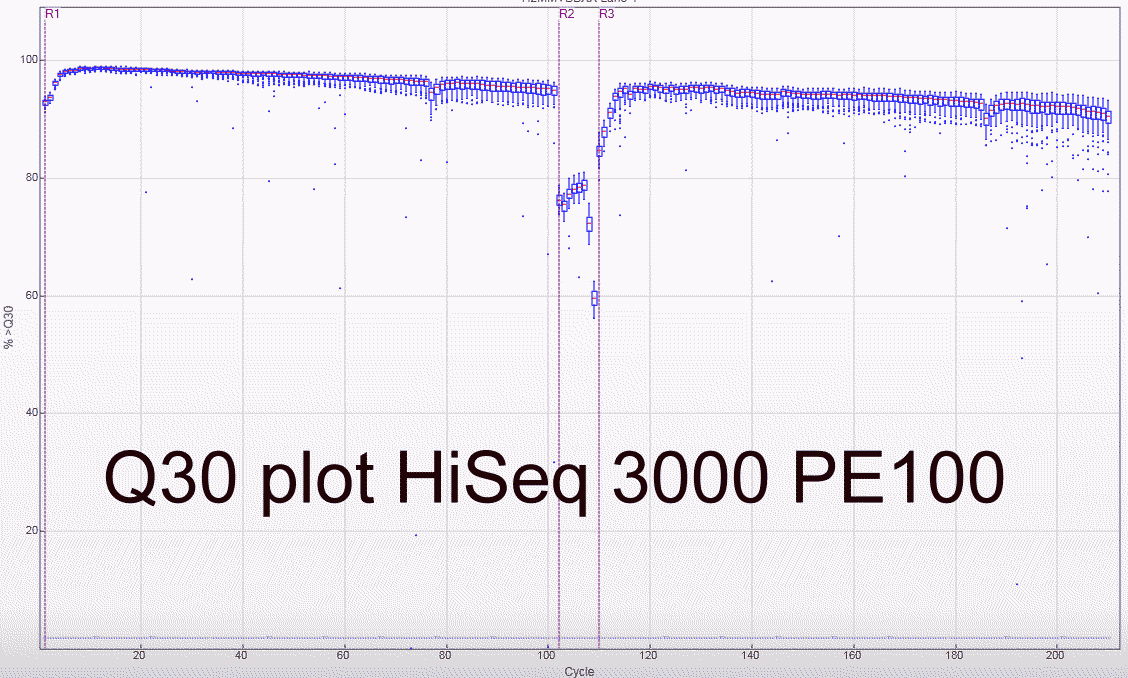

HiSeq3000/HiSeq4000 Libraries
- The new exclusion amplification on the flowcells more strongly advantages short library molecules (especially the even shorter adapter dimers) as compared to the previous bridge amplification protocol. According to Illumina 1% adapter dimer content can result in more than 6% of adapter dimer reads; 10% adapter dimer content resulted in 84% being unusable adapter dimer reads.
- Low library concentrations can lead to larger number of duplicates, even from PCR-free libraries, (likely the clusters are swapping over into neighboring cells).
- The minimum library concentration has increased to 2 nM.
- low complexity libraries
- bisulfite sequencing libraries
- even traces of adapter dimers
- Nextera libraries (no information from Illumina about the sequencing primers)
- any libraries with a considerable percentage of fragments with insert sizes over 550bp
- libraries with single-end adapters (these adapters should have been dropped years ago, though)
BTW, …
The cluster images do not give away the patterned character of the flowcells (click on the image to enlarge it).
