
Please also see the: Introduction to PacBio Sequencing and General Information or submit a sample with our submission form.
PacBio Sequencing Services and Performance
We offer complete PacBio sequencing services including HMW-DNA isolation, PacBio HiFi or Iso-seq library prep, size selection, and sequencing. Some secondary analyses HGAP assembly (small genomes), and IsoSeq analyses are included in the service. We will provide you with the complete data set generated by the PacBio sequencer and SMRT-LINK analyses for download from our servers. The Bioinformatics Core offers in-depth and custom analyses of PacBio sequence data. Due to the “single-molecule sequencing” technology, the PacBio sequence data quality and yields will depend highly on the quality of the DNA and RNA samples.
The PacBio Revio and Sequel II Sequencers
The Revio represents the fourth generation of PacBio sequencers. It significantly reduces the costs of long-read sequencing. While the principles of the PacBio sequencing chemistry remain, the advances and changes with the Revio are:
- The Revio increases the yield per SMRT-cell three times and increases the throughput 15 times compared to the predecessor Sequel IIe.
- The Revio is dedicated to high-fidelity (HiFi) long reads sequencing (CLR reads are not supported)
- HiFi reads will have read lengths of ~15 kb to 18 kb at a quality of Q30 or better (99.9% accuracy).
- HiFi data yields should range between 60 Gb and 90 Gb.
- Data are processed on the sequencer. No raw data will be delivered. The default data format will be bam files, including many metadata. You will receive mainly two files HiFi-reads (the circular consensus sequences) and fail-reads. Data can also be delivered in FASTA and FASTQ formats upon request.
- Epigenetics: DNA sample read data can include the 5mC methylation information.
- For whole genome sequencing please submit at least 5 microgram of high-quality DNA. Generally, this will be sufficient to generate a sequencing library for one Revio SMRT-cell.
- Full-length sequencing of 10X Genomics single-cell RNA-seq libraries is supported via the MAS-seq kit and protocol.
- The Revio currently requires library molecules longer than 3 kb. Thus shorter amplicons as well as Iso-seq libraries should be sequenced on the Sequel II sequencer.
- To make full use of the Revio, PacBio will soon be launching MAS-seq kits for amplicons and Iso-seq. This protocol concatenates shorter molecules in a controlled fashion and will enhance the data yield five times or more.
- Please see the information from PacBio on the new sequencer.
- This page summarizes the most frequent applications for Sequel sequencing as well as the expected required sequencing efforts (number of SMRT-cells required).
HiFi Sequencing: Unlike other technologies, PacBio sequencing adapters have hairpin structures. This allows the repeated circular sequencing of both strands of the library molecules. Read errors are efficiently removed from the sequences via Circular-Consensus-Sequencing (CCS) analysis, resulting in the highest sequencing data quality currently possible. Sequencing four passes of a library molecule is expected to yield Q20 data and nine or more passes Q30 data (99.9% accuracy). In contrast to other technologies, PacBio sequencing chemistry is not sensitive to extreme GC contents. Even long GCC repeat stretches can be sequenced. Please see these slides for more details.
Most Common Applications for PacBio Sequencing
Whole Genome Shotgun Sequencing: With the long & accurate HiFi reads PacBio sequencing is currently the default protocol for de novo genome assemblies. Three different library preparation protocols are available accommodating a wide range of sample input amounts: Standard HiFi libraries, Low Input libraries, and ultra-low input libraries.
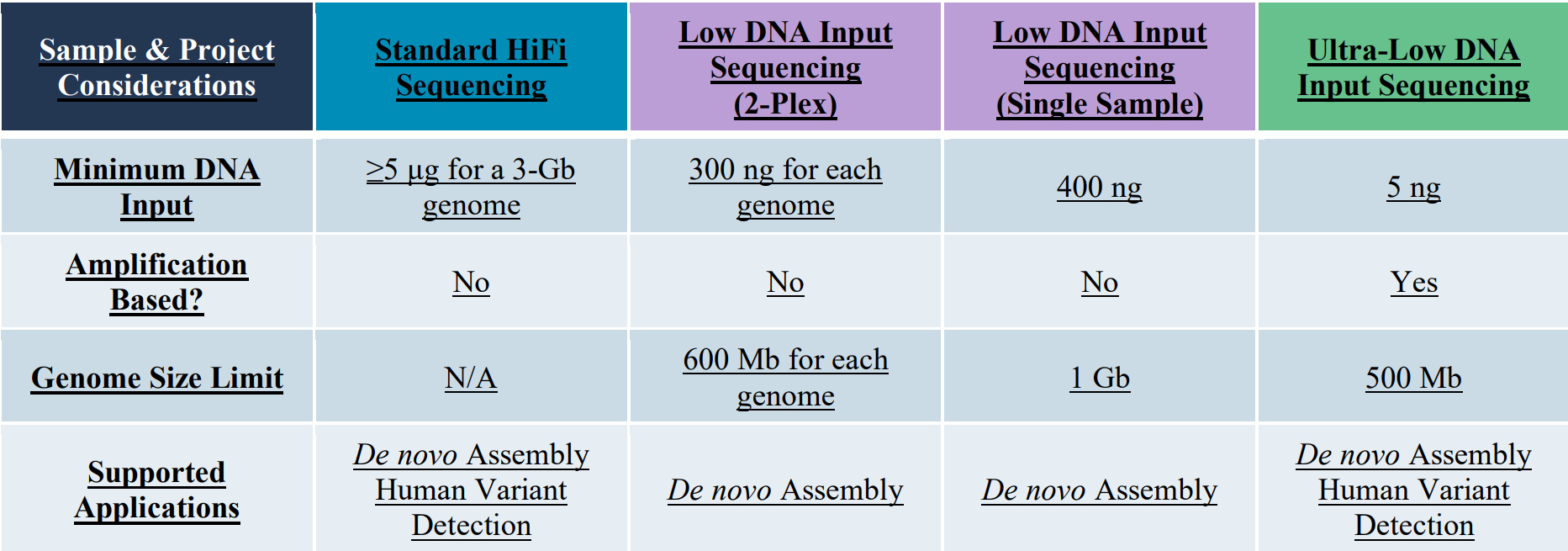
Please note that greater HMW-DNA sample amounts than those listed in this table, will improve the processing options and are certainly preferred. Typically, a genome coverage of about 20X to 30X is used for de novo assemblies. This is a generalization and experiment will need to be adjusted based on heterozygosity, ploidy, DNA quality, repeat content, etc. . Similarly simplified the sequencing of one Revio SMRT-cell can be expected to be sufficient for the assembly of a 3 Gb-sized genome. The PacBio data is also used to identify structure genomic variants at a coverage of ~10X.
For bacterial genome sequencing, the PacBio data can be analyzed for DNA methylation (the m6A and m4C modifications in prokaryotic DNA).
Iso-Seq (full-length transcript RNA-Seq): The PacBio long reads enable sequencing of full-length transcripts up to 10 kb (essentially all transcripts), thus eliminating the need for error-prone transcript assembly from short treads. The Iso-Seq bioinformatics pipeline processes the data into high-quality consensus transcript sequences enabling accurate isoform annotation and open reading frame prediction. These features do make Iso-Seq the method of choice for example for de novo gene annotations. Iso-Seq libraries can be barcoded and pooled at different stages of the library prep (after the reverse transcription; after PCR-amplification of cDNAs; after PacBio library prep). Please note that the transcript lengths can vary significantly between tissues and sample types. Thus, the read numbers per sample cannot be predicted accurately and the read numbers can vary significantly between pooled Iso-Seq libraries. Pooling the samples earlier in the library prep process will result in lower costs but also in higher variation of read numbers.
Long Amplicon Sequencing: Please see our FAQ for details. Amplicons of up to 15 kb length can be sequenced for highest quality CCS data. We offer two sets of universal barcodes PCR primer sets (12×12 and 24×24 indices) that you can pick up in the lab (for a fee). These allow the pooling of up to 576 amplicon samples.
The high-fidelity long read approach (10kb to 20 kb CCS sequencing with 30 hour runs) enables hihgest accuracy genome assemblies, haplotype phasing, the analysis of metagenomes, population samples, as well as the assembly of polyploid and highly heterozygous genomes.
Sample Requirements, Sample Submission and Scheduling
PacBio library prep requires microgram DNA sample amounts. Since the PacBio technology interrogates single molecules, any defect (e.g. a nick, an abasic site, a DNA adduct) can interfere with the sequencing process. Thus, the integrity and purity of the DNA sample is of utmost importance. The DNA quality and the DNA amount will determine which library insert sizes are feasible and how many SMRT-cells can be sequenced. The DNA samples should fulfill these criteria:
- Minimal DNA purity: OD 260/280 should be 1.8-2.0; OD 260/230 should be >2.0
- Has undergone a minimum of freeze-thaw cycles.
- Has not been exposed to high temps (> 65°C for more than one hour can cause a detectable decrease in sequence quality).
- Has not been exposed to pH extremes (< 6 or > 9).
- Does not contain insoluble material.
- Is RNA-free.
- Has not been exposed to intercalating fluorescent dyes or ultraviolet radiation.
- Does not contain, divalent metal cations (e.g., Mg2+), denaturants (e.g., guanidinium salts, phenol), or detergents (e.g., heme, humic acid, polyphenols). DNA samples should be submitted in EB buffer (EDTA-free) or TLE buffer (10 mM Tris, 0.1 mM EDTA, pH= 8 to 8.4).
- Microbial DNA samples must be isolated with silica spin column protocols (e.g. Qiagen DNeasy)
- All DNA must be double-stranded. Single-stranded DNA cannot be converted into SMRTcell templates but can interfere with polymerase binding.
The sample requirements will vary strongly depending on genome size. Please contact us to discuss your project.
We do offer HMW-DNA isolation as a service.
DNA isolation protocols are listed here: Extractdnaforpacbio.com
One recommeded protocol for plant samples is the Inglis et al. sorbitol wash method. Please be aware that some sample type will require the testing of several HMW-DNA isolation protocols.
One HiFi library prep reaction will be good for sequencing a single or at most two Revio SMRT-cells. As with other single-molecule sequencing technologies, the read lengths and the sequencing yields do depend on the nucleic acid sample quality. Every nick or chemical DNA adduct has the potential to abort a read. For difficult DNA samples, especially plant DNA samples with hard-to-remove contaminants (e.g. some polysaccharides), we recommend carrying out a high-salt/phenol/chloroform cleanup (please note that this protocol often leads to a loss of 50% of the sample). We will QC your sample by Pulsed-Field Gel Electrophoresis before library prep (PFGE; on BioRad CHEF, Pippin Pulse) and spectrometry. A band (not a smear) of 50 kb or longer fragments indicates high-integrity DNA samples desired for the generation of long insert size libraries. Please note that spectrometry and PFGE can not fully assess the quality and suitability of the DNA samples since they asses the DNA as double-stranded molecules. For example single-strand nicks and chemical adducts could escape these methods. The QC data however allow us to rule out clearly problematic samples.
The final quality assessment of the DNA sample will however be the single molecule sequencing process itself (e.g the average read lengths). Bacterial DNA samples extracted using silica columns will be sheared by the spin columns to fragments of about 15 to 20 kb in size. Such bacterial samples tend to generate high-quality data and are acceptable. Bacterial DNA samples must be isolated with silica spin column protocols (e.g. Qiagen DNAeasy) for submission to our core.
Before submitting samples:
- Please email us a picture of an agarose gel of the sample running also a marker with at least a 20 kb upper band (e.g. GeneRuler 1 kb Plus DNA Ladder or Lambda DNA/HindIII Digest Marker; suggested is a also a lane with undigested Lamdba phage DNA [48kb e.g. NEB N3011S]). Please run the electrophoresis slowly (e.g. at 80V depending on setup).
- Assess sample purity via spectrophotometry – the 260/280 ratio should be between 1.8 to 2.0 and the 260/230 ratio should be higher than 2.0. PacBio recommends MoBio PowerClean columns for sample cleanup or the high-salt/phenol/chloroform protocol mentioned above if necessary.
- Please use fluorometric methods (e.g. Qubit) for DNA quantification if possible. Measure each sample at least 3 times and accept only reproducible measurements (HMW DNA is often not perfectly dissolved). Spectrophotometry is not reliable for quantifications (especially if the DNA extraction protocol used CTAB).
The DNA samples used for making PacBio libraries must be handled with extreme care – if you need to ship your DNA to our facility, please consult the following PacBio guidelines for shipping and handling. More info and the submission form can be found here. We must receive electronic and print copies of the submission form.
Iso-Seq (Pacbio RNA-sequencing) sample requirements:
The RNA for IsoSeq experiments needs to be of high integrity (RIN-score 8 or better) and the Nanodrop 260/230nm ratio should be higher than 1.5 and the 260/280nm ratio between 1.8 and 2.1. The samples should best have a concentration of 50 ng/ul or more. 1 ug total RNA per sample will be sufficient, but more is always better.
Prices
Costs for PacBio sequencing reflect the number of libraries and number of SMRT cells required. Our recharge rates can be viewed here. The listed fees include all labor and reagents.