
 We are sequencing Illumina-style libraries on both Illumina and Element Biosciences AVITI sequencers. While the sequencing chemistries are very different, the general library requirements for both types of sequencers are very similar. The data format of the resulting sequences is also identical for all systems. Each sequencer offers a different sweet-spot when considering the main criteria of speed, accuracy, cost per base or per read, and cost per run. We are happy to discuss the benefits on a call.
We are sequencing Illumina-style libraries on both Illumina and Element Biosciences AVITI sequencers. While the sequencing chemistries are very different, the general library requirements for both types of sequencers are very similar. The data format of the resulting sequences is also identical for all systems. Each sequencer offers a different sweet-spot when considering the main criteria of speed, accuracy, cost per base or per read, and cost per run. We are happy to discuss the benefits on a call.
Compared to the long-time standard – Illumina sequencers – the main AVITI differences are:
- the AVITI generates data with higher quality scores (most bases will have a Phred scale Q-score of 44; equivalent to less than one error in 10,000 bases)
- the AVITI tolerates longer insert sequencing libraries
- there is no index-hopping on the AVITI sequencer
- clustering-related read-duplication rates are much lower (non-detectable) compared to NovaSeq X (>10%)
- the AVITI requires higher sequencing library concentrations; these are still easy to achieve, especially when pooling several libraries
- the AVITI is compatible with standard Illumina libraries with TruSeq-style or Nextera-style adapter sequences. This applies to the vast majority of Illumina libraries. Small RNA-seq libraries will require custom sequencing primers on the AVITI. Not compatible are sequencing libraries with IDT/SwiftBio “Normalase” treatment.
Please also see the general information on Illumina sequencing as well as the comprehensive Sample/Library Requirements Table.
Library Requirements for Illumina & AVITI Sequencing
Sequencing libraries should be quantified by a fluorometric method (e.g. Qubit, PicoGreen) or by qPCR. If a spectrophotometer (e.g. Nanodrop) is used, we suggest submitting twice the requested amount of sample since this type of measurement is often unreliable. We will re-quantify all library pools before loading them onto the sequencers. In some cases the MiSeq sequencers can be loaded with lower library concentrations – please inquire with us.
Libraries should be submitted with Bioanalyzer traces (or equivalent). We can run the Bioanalyzer QC for you for a fee. The preferred buffer for sequencing libraries is EBT (10 mM TRIS, 0.1% Tween20) or EB buffer (10 mM TRIS pH= 8.0-8.4).
The expected yield metrics apply to standard balanced libraries like RNA-seq or genomic libraries i.e., high-complexity libraries with average GC contents and of the recommended library molecule lengths. For these libraries you can expect yields that will exceed Illumina specifications; please see this FAQ.
The standard requirements for Illumina library submission are at least 15 ul volume at a concentration of 5 nM (e.g., 1.6 ng/ul given a 500 bp library) and 30 ul library at 16 nM concentration for the AVITI. More volume and/or higher concentrations are welcome. If required, 20 ul of custom sequencing primers need to be submitted together with the libraries at a concentration of 100 uM each. We can work with less library (down to 1 nM) for the MiSeq. Please note, we cannot guarantee the data quantity or quality for such low-concentration libraries as quantification becomes less reproducible, libraries become less stable, and relatively larger amounts of library DNA do stick to the sides of the storage tube. The best buffer to store and submit libraries is EBT (10 mM Tris/0.01% Tween-20 pH=8.0 or 8.4), but EB buffer is also acceptable. Please use 1.5 ml low-bind tubes (e.g. Eppendorf LoBind). If you do not provide a Bioanalyzer trace (or equivalent) of your library, we will run the Bioanalyzer for a fee. Please note, the DNA insert size(s) should not exceed 700 bp and most Illumina adapters add about 120 bases to the fragment length as observed on the Bioanalyzer. When submitting your libraries for sequencing, please use our Illumina/AVITI Sequencing Submission Form (hard copy with samples, and email to dnatech@ucdavis.edu), provide the Bioanalyzer profile, library prep methods, and index sequences used. We will measure the concentration of your libraries using real-time PCR (included in the sequencing price).
Libraries for NovaSeq sequencing – The latest generation of sequencers has more stringent library requirements and requires higher library concentrations. For the NovaSeq, average fragment lengths should ideally be between 420 bp and 700 bp and the “tail” of longer fragments should not exceed 1 kb. The longer library molecules will result in lower quality reads for this fraction of the samples and will impact read numbers reducing the yields. The new clustering chemistry is more sensitive to adapter dimers: a 5% adapter-dimer contamination can result in 60% of the reads coming from these dimers. Thus, it is very important that there is no indication of an adapter-dimer peak (around 120 bp) on the Bioanalyzer trace. Our preferred requirements for library submission for Illumina sequencing are for at least 15 ul volume of 5 nM concentration library or pool per run or lane (e.g., 1.6 ng/ul given a 500 bp library). More volume and/or higher concentration is welcome. Lower sequencing yield is the likely outcome for library concentrations 2 nM or less, and we cannot guarantee the data quantity or quality for such libraries.
We highly recommend using uniquely-dual-indexed (UDI) adapters for both the HiSeq and the NovaSeq. Matching i5 and i7 indices per library must be avoided. The UDI adapters allow the efficient elimination of all potential index hopping artifacts.
Demultiplexing is included with our sequencing services, however, please ensure your sample IDs and barcodes are unique; we are now implementing additional labor charges for re-demultiplexing because of incorrect submission info, duplicate sample names, wrong barcodes given, reverse complements given in error, etc.
Library Pooling Service – We offer the pooling of sequencing libraries for a fee. Prerequisites for the pooling of customer libraries are:
- all libraries were generated using the same protocol and are PCR amplified
- their fragment size distribution is similar (and within the Illumina specs) as demonstrated by Bioanalyzer traces
- uniquely indexed adapters
- all libraries need to have DNA concentrations in about the same range
Library pooling requires precise pipetting of very small volumes and we can’t work magic with wildly variable samples. PCR-free libraries are best quantified by qPCR. Other libraries can be quantified by fluorometry (e.g., Qubit). For sequencing libraries generated by our Core lab, pooling is included in the service. Please also see our FAQ.
PCR-Free Libraries – special QC required! The quality of unamplified libraries is difficult to assess. The adapters of these libraries are partly single-stranded. When assaying them via capillary electrophoresis they tend to migrate slower than the fully double-stranded, amplified libraries. In most cases, the libraries appear to be 70 to 100 nt longer than they actually are – however, the Bioanalyzer traces can also be off by far larger margins (e.g., 500 bases). To be sure about the actual library fragment lengths we ask you to PCR-amplify an aliquot (1 ul) of the libraries with 8 PCR cycles and run both the PCR-free and the amplified sample on the Bioanalyzer. If multiple PCR-free libraries will be pooled, you might consider quantifying the individual libraries by qPCR before pooling.
Libraries: Quality Control (QC)
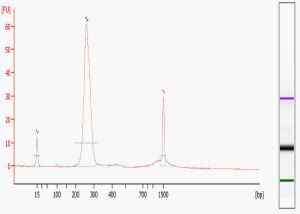 Library quality is the single most important determinant of the success of your sequencing run, both in terms of the number of reads generated (quantification) and the validity of the sequence obtained (content). We carry out two QC measures on all libraries sequenced in our Core – examination on the Agilent Bioanalyzer, and quantification using the Kapa Biosystems Illumina Library Quantification Kit. The “perfect” library Bioanalyzer trace, pictured here, shows a single peak of the expected molecular weight. Common additional forms include primer dimers (at around 80-85 bases), adapter dimers (around 120 bases), and broader bands of higher MW than the expected peak. Primer dimers are not a problem unless they completely dominate the reaction. Adapter dimers can be a problem because they will sequence much more efficiently than the longer library fragments. As a result, the proportion of adapter dimers in your library will be seen as an even higher percentage of reads in your final data files. Adapter dimers can be minimized by adjusting the adapter/insert ratio during library construction and exercising care in gel extraction, bead purification, or other size selection steps. The larger MW, typically more hump-shaped forms that are visualized on the Bioanalyzer are probably a result of excess amplification during the final PCR step. While some amount of these is tolerable, if they are too prominent then the library should be re-amplified from the gel-extracted material.
Library quality is the single most important determinant of the success of your sequencing run, both in terms of the number of reads generated (quantification) and the validity of the sequence obtained (content). We carry out two QC measures on all libraries sequenced in our Core – examination on the Agilent Bioanalyzer, and quantification using the Kapa Biosystems Illumina Library Quantification Kit. The “perfect” library Bioanalyzer trace, pictured here, shows a single peak of the expected molecular weight. Common additional forms include primer dimers (at around 80-85 bases), adapter dimers (around 120 bases), and broader bands of higher MW than the expected peak. Primer dimers are not a problem unless they completely dominate the reaction. Adapter dimers can be a problem because they will sequence much more efficiently than the longer library fragments. As a result, the proportion of adapter dimers in your library will be seen as an even higher percentage of reads in your final data files. Adapter dimers can be minimized by adjusting the adapter/insert ratio during library construction and exercising care in gel extraction, bead purification, or other size selection steps. The larger MW, typically more hump-shaped forms that are visualized on the Bioanalyzer are probably a result of excess amplification during the final PCR step. While some amount of these is tolerable, if they are too prominent then the library should be re-amplified from the gel-extracted material.
The electropherogram of a sequencing library should show a distinct peak. The tighter the library fragment size distribution is, the more accurately the library can be loaded onto the flowcells.
Library QC for PCR-free Sequencing Libraries PCR-free libraries pose special problems since they can show several types of misleading artifacts on Bioanalyzer runs. Thus we require a Bioanalyzer trace of the library to be sequenced, as well as a trace of an amplified aliquot of the same library. Please see this section of our FAQ for details.
Custom Sequencing Primers Please note that these are used only for a small minority of sequencing projects. Custom sequencing primers need to be submitted at a concentration of 100 uM and a volume of 20 ul each together with the libraries. Please make sure that the sequencing primer design fits the chosen Illumina platform; MiSeq and HiSeq platforms use different annealing temperatures.
Submission
All customer-submitted libraries should be accompanied by Bioanalyzer (or similar) traces. If no traces are submitted we will carry out the Bioanalyzer analysis for a fee. Please visit the Sample Submission & Scheduling page to download submission forms and for detailed instructions.
Scheduling
Once your library, or library raw material, is ready, you should deliver it as soon as possible to get the next available slot in the queue. Runs occur as we fill up flow cells, and the timing on this can vary depending on service type and Core activity. For MiSeq runs the turnaround time is typically five to ten days, while you should allow two to four weeks for NovaSeq sequencing; allow an extra eight to ten weeks for library prep if requested.
Sample/Library Storage Policy
Please let us know if you would like to pick up your samples/libraries after they have been sequenced and we will be happy to accommodate you; otherwise, due to space limitations, they will be stored for only six months after sequencing runs have been completed.