
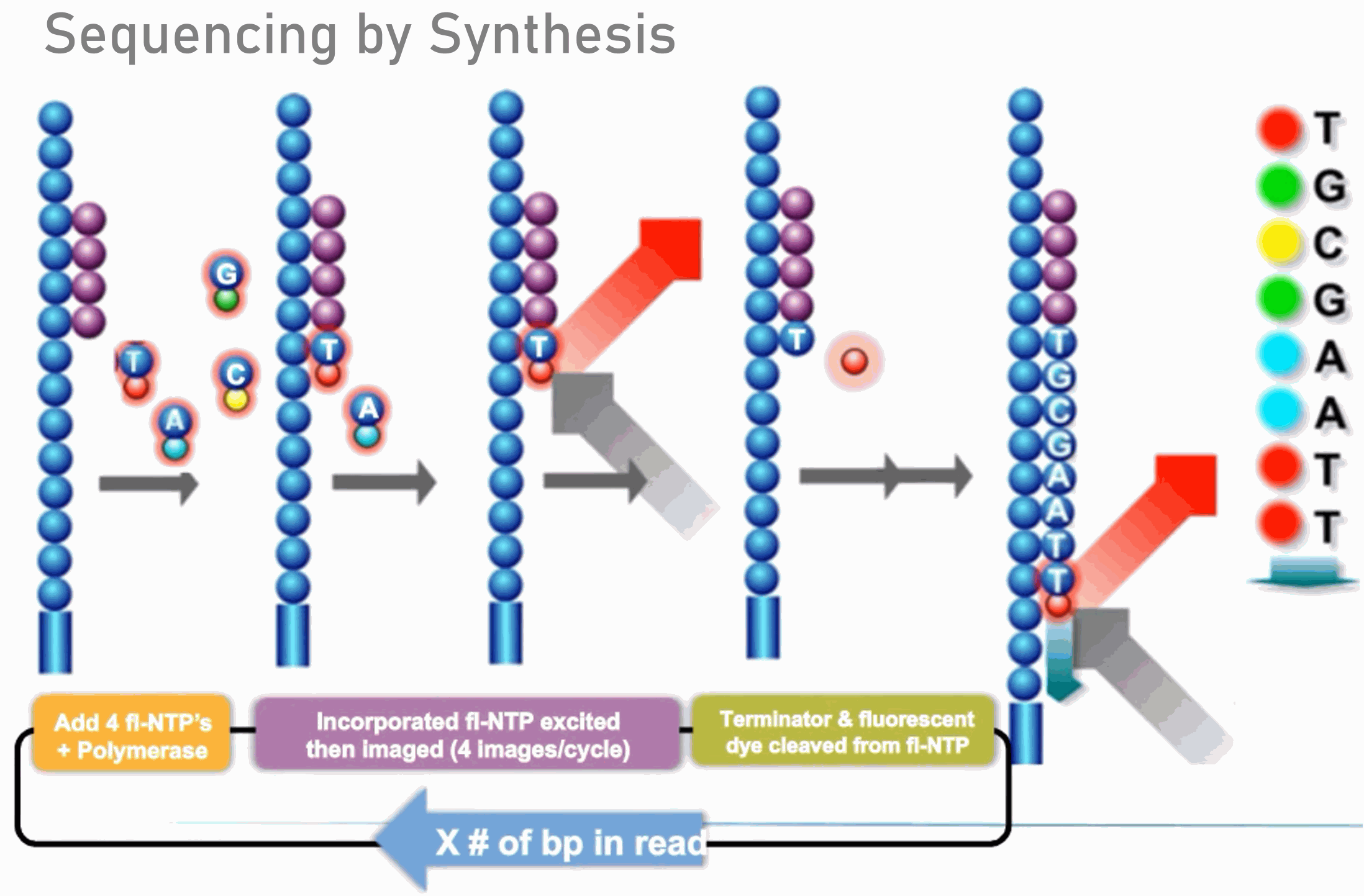
Illumina sequencing platforms generate up to 800 gigabases of high-quality sequence data per lane (NovaSeq 6000 S4) using a massively parallel short-read sequencing approach. The Illumina systems use sequencing-by-synthesis technology and reversible terminator chemistry. More information describing the technology and the instrument is available from Illumina here.
Getting started info: > Register with the GC > Sample Requirements > Sample Submission
The DNA Technologies and Expression Analysis Cores of the Genome Center operate one Nextseq 500, and three MiSeqs. We are also sequencing on the NovaSeq 6000.
Steps involved in an Illumina sequencing experiment can be broken down into a series of experimental manipulations, instrument runs, and data analyses. These steps include creation of sequencing libraries, seeding clustering of the flow cell on the sequencer, sequencing by synthesis, and bioinformatics.
Please also see the “Beginner’s Guide to NGS” information and the video at the bottom of this page, both from Illumina.
Read Length – Data Output
Current average short-read sequencing data yields:
When we received our first Illumina Genome Analyzer in August 2007, accurate read lengths barely exceeded 25 bases. Now we are getting excellent (i.e. low) error rates up to 150 bases of paired-end reads for NovaSeq 6000, or 300 bases of paired-end reads for MiSeq. The read length you require will depend on your experimental needs, and discussions with us and the Bioinformatics Core will be essential to focus your decision. Here are a few general guidelines:
A single read 100 bp (SR100 in Core lingo), paired-end 100 bp (PE100), paired-end 150 bp (PE150), rapid-mode paired-end 250bp (PE250 ), and paired-end 300bp (PE300 Miseq only) are standard read lengths offered by the Core. SR50 will already provide an unambiguous match to a reference genome and is therefore suitable for most ChIP-seq and mRNA-seq applications where a reference genome is available. SR100 is the read length used for small RNA and micro RNA analyses. It can also be used for certain SNP discovery applications, where a reference genome is available. It can be suitable for some bacterial genome analyses, and reduced representation or hybrid-selected libraries, depending on the application. The MiSeq generates about 12-15 million reads passing filter (using v2 chemistry) or up to 25 million reads PF (v3 chemistry). The following kits are available: SR50 (v2 kit), PE75 (v3), PE150 (v2), PE250 (v2), and PE300 (v3 kit). Considering the fast turnaround time (1-3 days once libraries are loaded for sequencing), this would be an excellent option for small-scale and proof-of-concept projects under a strict timeline.
 Paired-end reads substantially facilitate assemblies of all genome sizes. Paired-end reads can also help resolve differences among repeat regions and thus can be used in transcriptome projects to distinguish family members as well as identify alternative splicing. The most commonly run paired-end reads in the Core are PE100 and PE150 (HiSeq), and PE250 (Miseq and Rapid Mode Hiseq), and PE300 (MiSeq). For de novo sequencing, PacBio RSII is an excellent alternative or complementary option as this generates average median read lengths of > 10kb with long reads tailing off around 40kb. Please see more details on PacBio sequencing here.
Paired-end reads substantially facilitate assemblies of all genome sizes. Paired-end reads can also help resolve differences among repeat regions and thus can be used in transcriptome projects to distinguish family members as well as identify alternative splicing. The most commonly run paired-end reads in the Core are PE100 and PE150 (HiSeq), and PE250 (Miseq and Rapid Mode Hiseq), and PE300 (MiSeq). For de novo sequencing, PacBio RSII is an excellent alternative or complementary option as this generates average median read lengths of > 10kb with long reads tailing off around 40kb. Please see more details on PacBio sequencing here.
Did it Work?
The question of what defines a “good” sequence is of some interest. Every sequencing lane run contains some control phiX174 DNA. We determine whether a run meets quality specifications based on the behavior of this commercially available library DNA. This is evidenced by certain metrics: percent of clusters that pass quality filtering (PF), number of sequences that align to the phiX reference genome, and the overall percent error rate for those aligning phiX sequences. The summary report of the run (in a file called Summary.htm) is available on SLIMS so you can see these metrics on the control for yourself. Depending on your library and the quality of the reference genome used in your alignment you may not have access to these same metrics, so the behavior of the control lane can be informative. Even if you don’t do an alignment or have a good reference genome there are other indicators of quality. At the gross level there is base composition which tells you, for example, whether the genome you sequenced matches the genome of your organism, or whether there’s a preponderance of linker/adapter sequences in your output. The Summary.htm file reports the percent of your clusters that pass filtering, which is indicative of the overall quality of the run (but unfiltered reads may still contain good data). At a deeper level, quality scores for each base in each read are contained within some of the files themselves (see the pipeline manual for more info), although parsing the data at this level requires more bioinformatics knowledge.
Prices
Prices for sequencing depend on the number of cycles requested (i.e., how long a sequence you want) and whether the runs are single read or paired-end. However, additional options and services are continually becoming available, so for the latest prices and offered services please contact Lutz Froenicke or Vanessa Rashbrook. The listed fees include all the labor and reagents for cluster generation, cycle sequencing, and initial base calling analysis using the standard Illumina pipeline and sample de-multiplexing according to index read data. Failed lanes have become extremely rare on the latest equipment. Even from sub-par runs some information, such as library integrity, can be obtained. If re-runs are necessary they will be carried out as soon as possible and not moved to the end of the queue. While initial base calling and data demultiplexing (from index reads) is included in our services, the Bioinformatics Core has developed a menu of services relating to the access, manipulation and analysis of sequence data from these instruments. Please contact them for help in analyzing your data.
Sample Submission and Scheduling
We must receive electronic and print copies of the appropriate submission form in order to receive and schedule your samples. Please visit the Sample Submission & Scheduling page to download forms and for more details. Please see the HiSeq Sequencing Calendar and Queue for the current HiSeq3000 and HiSeq2500 schedules.
Custom Sequencing Primers
Custom sequencing primers can be used if provided (the custom primer’s Tm should match that of the illumina primer, since amplification is isothermal). Please note that the sequencing primer requirements do vary between IIlumina platforms and chemistries (e.g. the Miseq tends to require higher melting temperatures). Custom sequencing primers are not supported by Illumina. Nevertheless please discuss the primer requirements with the Illumina tech support. If custom sequencing primers are employed the user takes full responsibility for the design; we can not vouch for the success of sequencing runs with custom primers.