As of January 2020, 10X Genomics has announced that it is discontinuing production of the 10X Chromium Genome linked read reagents. We are sad to see this very popular assay go.
Update July 2020: We have ~16 remaining 10X Chromium Genome reactions left in our core. As soon as these run out, we will discontinue the assay effective immediately. Please contact dnatech@ucdavis.edu to reserve reagents for your project.
We are offering 10X Genomics Chromium Genome “linked read” library preps and sequencing. The sequencing libraries are generated with the Chromium controller instrument and reagents from 10X Genomics, followed by sequencing on our Illumina HiSeq sequencers. When analyzing high molecular weight DNA samples the linked read sequencing data delineate linkage information over distances of up to 150 kb. Just a few nanograms of sample ar e required as input for the library preparation.
e required as input for the library preparation.
The applications currently supported by the 10X Genomics software are human haplotype phasing and structural variant detection (see this page for currently supported applications) as well as de novo genome assemblies (please see below). Genotyping in repetitive regions of genomes is possible via an approach called “critical content rescue”. Please see these slides for details on GemCode technology and some of the possible applications.
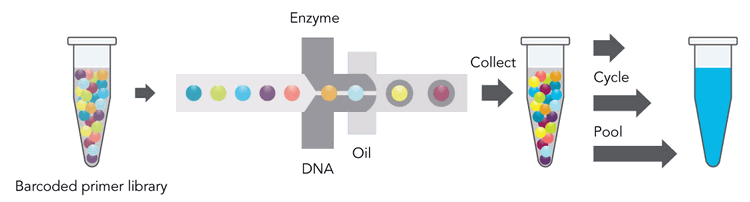
The 10X Genomics technology generates individually barcoded sequencing libraries for hundreds of thousands of nanoliter volume oil droplets using up to 1.7 million different barcodes. Thus, it allows individual long DNA molecules 10X Genomics Chromium Genome Linked-reads principle. DNA equivalent to about 150 copies of a genome does get distributed over about 1 million oil droplets that include beads. Sub-genome sized parts of the DNA sample (about 1/6000th of a genome) are compartmentalized in nanoliter volume oil droplets together with beads carrying one of about two million different barcodes to initiate the library preparation with droplet specific barcodes. Please note that the individual DNA molecules are not meant to be fully assembled with this approach. A single HiSeq lane generates sufficient information for phasing and structural variant analysis of a human sized genome. Human phasing can be carried out using both whole genome shotgun sequencing as well as employing exome capture enrichment. For human whole genome phasing and structural variant analysis GemCode sequencing data at 60x genome coverage or a combination of conventional Illumina data coverage at 30x genome coverage and GemCode at least 23x is recommended. The 10X Genome sequencing data contain conventional shotgun sequencing data, after trimming off the first 26 bases of the forward read.
The DNA quality will determine to a great extent the linkage information that can be gained from the 10X data – the longer the input DNA fragments the greater the length of the linkage information per droplet. 10X Genomics does suggest the Qiagen MagAttract HMW DNA Kit for DNA isolations (for plants other DNA extraction methods will be required). Please see this suggested protocol. The quality of the DNA samples should be verified by pulsed-field gel electrophoresis. The DNA amount required for the actual library prep is minimal (1 ng), however more DNA is required for the QC of the sample (we suggest to submit at least 500 ng at a concentration of 20 ng/ul or higher as determined by Qubit).
10X Genomics recommends DNA sample should run as a band of 50 Kb length or higher on a pulsed-field gel. For DNA samples at this size we might recommend two pre-treatments: DNA-Damage-Repair and BluePippin Sample Size Selection (the latter will remove fragments shorter than 40 kb). For DNA samples significantly longer than 50 kb these pre-treatments will likely not be necessary. The first step of the actual 10X library prep is a denaturation of the sample. Thus nicks present in the DNA sample will significantly reduce the linked-read length.
The 10X Genomics Chromium Genome assays do require paired-end 150 bp read sequencing. We offer the isolation of HMW-DNA samples – please see this page.
De novo Genome Assembly Projects: 10X Chromium sequencing data can yield excellent de novo genome assemblies when assembled with the free Supernova2 genome assembler. Please note that the 10X Genomics Chromium technology was designed primarily for the analysis of human genomes. Thus, the design is optimized for large genomes and works best for genome sizes around 3 Gb. However, the Bioinformatics Core has generated and high quality genome assemblies with scaffolds comprising entire chromosome arms for both birds and mammals from our 10X data. A bird genome assembly showed a contig N50 of 147 kb and a scaffold N50 of 18 Mb for a 1.2 Gb genome. The means the assembly metrics are limited by the karyotype comprising multiple micro-chromosomes. This assembly was based on a single 10X library and a single HiSeq lane of data.
The assembly results will vary significantly depending on DNA sample quality, genome size and genome organization (e.g. repeat content and structure). Please note that the Supernova genome assembler was designed only for diploid genomes. As in most cases plant genomes tend to be more difficult to assemble. In general, the results are better than other assemblies based on Illumina sequencing data (using shotgun and mate-pair data). Please see this link for the recommendations from 10X Genomics:
https://support.10xgenomics.com/de-novo-assembly/software/overview/latest/performance and these suggestions for input amounts and data sub-sampling (note that 10X counts forward and reverse reads individually). 10X Genomics has only tested the assembly of genomes of up to 3.4 Gb. Theoretically Supernova 2 could be able to assemble genomes of up to 5 Gb size.