
Nanopore sequencing with Oxford Nanopore Technologies (ONT) systems enables high-throughput long-read sequencing of both DNA and RNA samples as well as multiple base-modifications including 5mC and 5hmC. For highest molecular weight DNA (HMW-DNA) samples, read lengths of several hundred kb can be reached with ultra-long-read protocols. There is however a significant tradeoff between read lengths and expected sequence yield. Nanopore sequencing data greatly enable de novo genome assemblies and structural genomic variant and transcriptome studies. ONT flow cells accurately measure the current/electrons that squeeze by single-stranded DNA molecule as it is ratcheted through a nanopore molecule between two compartments. The signal of the changing current is converted to base-calls via machine learning algorithms. To learn more about the technology, please see the ONT HOW-IT-WORKS page and the video below.

We offer Nanopore sequencing on the highest throughput Nanopore sequencer, the PromethION. To our knowledge, we were the first academic core lab to offer such services. Depending on the samples and the library types, the PromethION can generate more than 100 Gb of sequence data per flow cell, and can run up to 24 flow cells simultaneously. We do offer HMW-DNA isolation as a service since high-quality single-molecule sequencing data depend on highest quality DNA samples.
Getting started info: > Register with the GC > Sample Requirements > Sample Submission
Nanopore technology is progressing especially rapidly and is characterized by roughly annual flowcell chemistry changes and even more frequent basecaller software updates, resulting in quickly improving sequence qualities.

Average sequence data yields can vary significantly and will mostly depend on the sample properties (please see below). Average yields will be inversely correlated to the library molecule lengths (insert sizes) in a range from 200 kb down to 20 kb. We have achieved the best run metrics mentioned below with DNA isolated from human/mammalian cell cultures and mammalian blood samples.
We offer different types of PromethION sequencing:
- Long-read DNA sequencing: of sheared DNA fragments from 5 kb to 50 kb. Yields per PromethION flowcell can range from 20 Gb up to 130 Gb.
- Super-long-read DNA sequencing: For this protocol, we shear the DNA to fragments of about 50 kb lengths using the Diagenode MegaRuptor and then ligate-on the sequencing adapters. The yields per flow cell can vary from 20 Gb to up to 90 Gb; read lengths N50 values can reach up to 33 kb and longest reads of up to 270 kb length.
- Ultra-long-read DNA sequencing: Here, the sequencing adapters are added by a transposase instead of a ligases. The goal is a significant proportion of reads with lengths longer than 100 kb. However, the yields are expected to be significantly lower compared to the super-long reads. This type of sequencing is already possible on the MinION sequencer where we have seen read lengths of up to 840 kb and read lengths N50 of up to 70 kb (MinION). It is expected that a combination of super-long-read and ultra-long read data will assist de novo genome assembly projects tremendously.
- Full length cDNA sequencing: The cDNAs will be prepared after priming from the poly-A tails (analogous to the Clontech SMART-seq protocol) and PCR amplified. The cDNA amplicons are converted to Nanopore libraries via adapter ligation. These data are used for isoform analysis and gene annotations. In contrast to the PacBio Iso-Seq protocol, the data cannot be processed for high-accuracy circular consensus information (CCS) though.
- direct RNA sequencing: this protocol is being implemented. The library preparation adds an adapter via annealing to the poly-A tail. The sequencing data can be used to investigate RNA-base modifications.
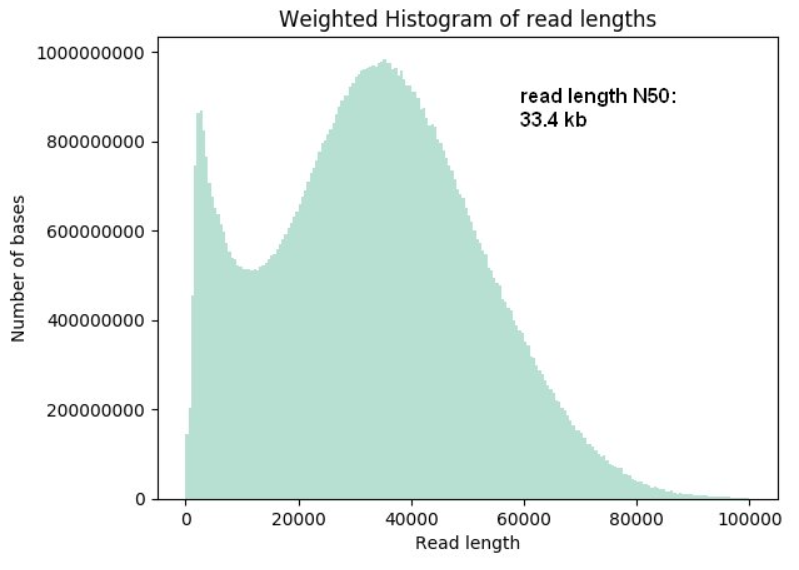
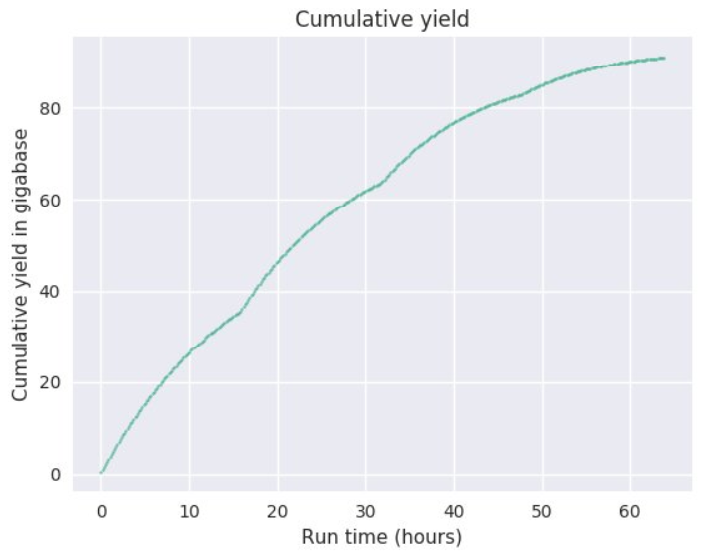
The sequence yields and run metrics will vary widely depending on the samples. As with other single-molecule sequencing technologies, the read lengths and the sequencing yields do depend on the nucleic acid sample quality. Every nick or chemical DNA adduct has the potential to abort reads and chemical contaminants could destroy the pores. The sample quality can be assessed in part by spectrometry and pulsed-field-gel-eletrophoresis (PFGE). We will QC the samples with both. Please note that these methods can not fully assess the quality and suitability of the DNA samples since they assess the DNA as double-stranded molecules. For example, single-strand nicks and chemical adducts could escape these methods. The QC data however allow us to rule out clearly problematic samples. It has been observed that genomic DNA samples from certain species (including some birds and Cnidaria) perform significantly worse than for example, mammalian samples. Thus it is possible that genomic DNAs of some species contain DNA base modifications that interfere with the current Nanopore protocols. As a consequence, we will perform test sequencing experiments (also on the smaller MinION sequencer) with new types of samples. Test sequencing is the only way to fully assess the suitability of DNA samples. We can’t make any specific sequencing performance promises beforehand.
It can be expected that Nanopore sequencing will generate on average longer reads as compared to sequencing on the Pacbio Revio. Please note, however, that Nanopore and PacBio data are not equivalent.
Our Nanopore sequencing expert is Ruta Sahasrabudhe, PhD. Please contact her at rmsaha@ucdavis.edu for questions about HMW DNA extractions, Nanopore Sequencing & Hi-C projects.
In contrast to other sequencing technologies, the PromethION sequencer is very robust, since it is purely an electronic system – the chemistry is confined to the disposable flowcells. Thus, UC Davis scientists can get trained to sequence on the PromethION themselves during downtime periods.
Sample requirements:
Genomic DNA samples:
DNA samples have to be chemically pure with Nanodrop spectrometer 260/280 nm ratios between 1.8-2 and 260/230 nm ratios between 2.0-2.2. The DNA needs to be dissolved in 10 mM TRIS (pH=8.0-8.4) – e.g. Qiagen EB Buffer.
For long-read sequencing genomic DNA samples isolated with spin column protocols ( e.g. Qiagen DNeasy) are sufficient. Please submit at least 5µg of total DNA at a concentration of 100ng/µl in 50µl volume. Please submit a gel image.
For super-long-read sequencing we require high molecular weight DNA samples with fragment sizes over 50KB. Please submit at least 5µg of total DNA at a concentration of 100ng/µl in 50µl volume. Please submit a gel image.
For difficult DNA samples, especially plant DNA samples with hard-to-remove contaminants (e.g. some polysaccharides), we recommend to carry out a high-salt/phenol/chloroform cleanup . Please note that this protocol often leads to a loss of 50% of the sample.
Before submitting DNA samples:
- Please email us a picture of an agarose gel of the sample. The run should contain also a marker of at least a 20 kb size (e.g. GeneRuler 1 kb Plus DNA Ladder or Lambda DNA/HindIII Digest Marker; suggested is a also a lane with undigested Lamdba phage DNA [48kb e.g. NEB N3011S]). Please run the electrophoresis slowly (e.g. at 80V depending on setup).
- Assess sample purity vie spectrophotometry – the 260/280 ratio should be between 1.8 to 2.0 and the 260/230 ratio should be higher than 2.0. PacBio recommends MoBio PowerClean columns for sample cleanup or the high-salt/phenol/chloroform protocol mentioned above if necessary.
- Please use fluorometric methods (e.g. Qubit) for DNA quantification if possible. Measure each sample at least 3 times and accept only reproducible measurements (HMW DNA is often not perfectly dissolved). Spectrophotometry is not reliable for quantifications (especially if the DNA extraction protocol used CTAB).
- The samples have to be RNA-free. The DNA isolation protocols should best include an RNAse digestion.
We do offer HMW-DNA isolation as a service.
Suitable DNA isolation protocols are listed here: Extractdnaforpacbio.com
HMW-DNA isolation and Nanopore sequencing from animal samples:
Cell culture samples: Please submit 10-50 million cells or more for HMW DNA extraction. Cells should be pelleted, washed with 1X PBS, supernatant removed, flash frozen in liquid nitrogen and stored at -80C. Please ship the cell pellets on dry ice.
Animal tissue samples:
Fresh tissue must be snap frozen in liquid nitrogen right after harvesting and stored at -80 0 C. Frozen sample should be shipped on dry ice. Any freeze-thaw cycles should be avoided. Please submit at least 500 mg of soft tissue; not including guts.
Blood samples:
Samples should be collected fresh with EDTA as anticoagulant. We prefer to receive fresh blood samples shipped overnight on cold packs; these samples should be stored in a refrigerator temporarily. Blood samples can also be flash frozen in liquid nitrogen and stored at -80 0 C. Frozen samples should be shipped on dry ice. About 5 mls of mammalian blood sample will be required each for HWM DNA isolation. Please contact us beforehand to set up an appointment if you plan to ship fresh blood samples with overnight shipping.
For the sequencing over a larger number of flowcells, additional material might be required.
RNA samples:
For full-length cDNA sequencing we require 1 ug of total RNA in up to 20 ul of molecular-biology-grade water. The RNA samples need to be DNA-free and need to be accompanied by Bioanalyzer traces. The samples should have RNA-integrity scores (RIN-scores) of 8 or higher.
Data delivery:
We will distribute Nanopore sequence data in fastq format via a secure server (Bioshare). The raw Nanopore sequence data in fast5 format are extremely large and unwieldy – each read is an individual file. We can distribute the raw data via hard drives if requested. We will transfer them to new portable hard drives and charge for the additional costs.
Nanopore Sequencing Technology:
ONT graphic showing the Nanopore (blue) embedded in a membrane. The motor protein (green) sitting on top of the pore unwinds the double helix and ratchets a single DNA strand through the pore. The data plot illustrates the raw signals: measurements of the electric current passing through the pore. The strength of the current is influenced mostly by a stretch of five DNA bases located in the bigger cavity of the pore. Due to the ratcheting motion of the motor enzyme each base contributes to five signals.