
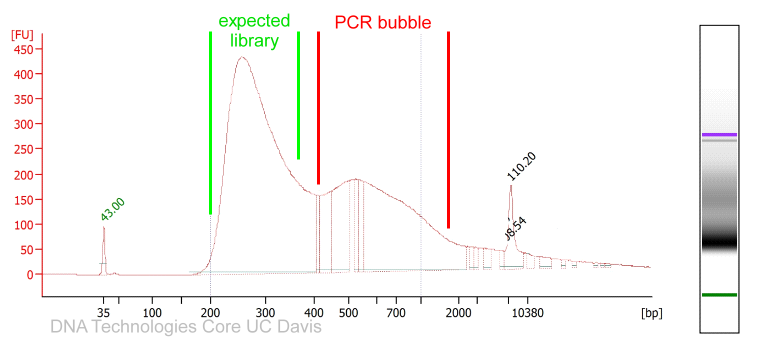
PCR amplified sequencing libraries frequently display library molecules seemingly about twice the excepted size or even bigger. In most cases, this phenomenon is caused by over-amplification of the libraries. These PCR artifacts do occur in cases the PCR reactions run out of essential reagents – in most cases the PCR primers will be exhausted. If primers are no longer available the PCR products will anneal to each other (the sequencing adapter sequence will be the by far most common sequences available). The resulting annealing products are often called “PCR-bubbles” and are partly double-stranded and partly single-stranded; thus they migrate considerably slower on agarose gels as well as on Bioanalyzer assays. Please see below.
Since these artifacts are merely annealing products, the resulting libraries are perfectly sequence-able. However, the quantification of such libraries by fluorometry will not be precise since the dyes used for these measurements are specific for double-stranded DNA molecules and PCR bubbles contain considerable amounts of single-stranded DNA that will not be measured. The PCR bubbles can be removed by amplifying the library one more time with a single cycle of PCR (a so-called “Reconditioning PCR“). For this PCR you could use standard Illumina P5 and P7 primers (please see below). The complementary sequences should be located at the very ends of all Illumina sequencing library molecules. In most cases PCR bubble artifacts can not be removed by SPRI bead size selections or Blue Pippin size selections; if necessary, a “Reconditioning PCR” is the best option.
However, to avoid unnecessary complexity loss of the library and introduction of polymerase errors, it would be best to optimize the library preparation protocol for a lower number of PCR cycles beforehand.
Reconditioning PCR protocol:
The reconditioning PCR uses standard Illumina P5 (5′-AATGATACGGCGACCACCGAGATCT-3′) and P7 (5′-CAAGCAGAAGACGGCATACGAGAT-3′) PCR primers which can be ordered as desalted DNA oligos.
Create a 10x concentrated primer mix at 20 μM each of these primers in EB buffer.
Add 1 to 4 μl previous PCR product, 2 μl 10x primer mix, 3 to 7 μl water for a total volume of 10 μl, then add 10 μl Kapa HiFi 2x Hotstart PCR master mix and pipette up and down several times.
Use the following cycling parameters: Initial denaturation 98°C 45 sec, denaturation 98°C 15 sec, annealing 60°C 30 sec, extension 72°C 30 sec, final extension 1 min.
Perform a standard Ampure XP/SPRI bead cleanup (e.g. with beads at 1.2x the sample volume) before analyzing the product by microcapillary electrophoresis.
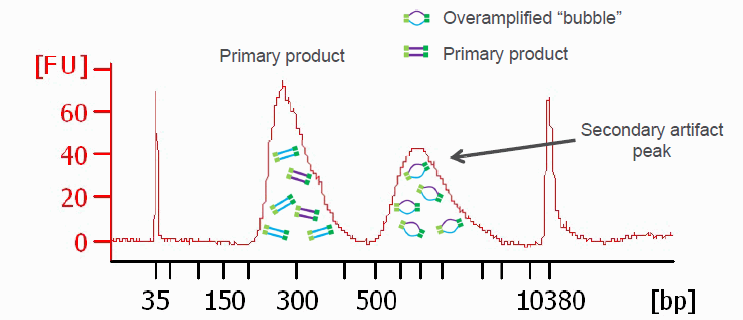
Another graphic illustrating PCR bubbles (source Illumina Inc.). Please also see: https://support.illumina.com/bulletins/2019/10/bubble-products-in-sequencing-libraries–causes–identification-.html
← FAQ