
UMI is an acronym for Unique Molecular Identifier. UMIs are complex indices added to sequencing libraries before any PCR amplification steps, enabling the accurate bioinformatic identification of PCR duplicates.
UMIs are also known as “Molecular Barcodes” or “Random Barcodes”. The idea seems to have been first implemented in an iCLIP protocol (König et al. 2010).
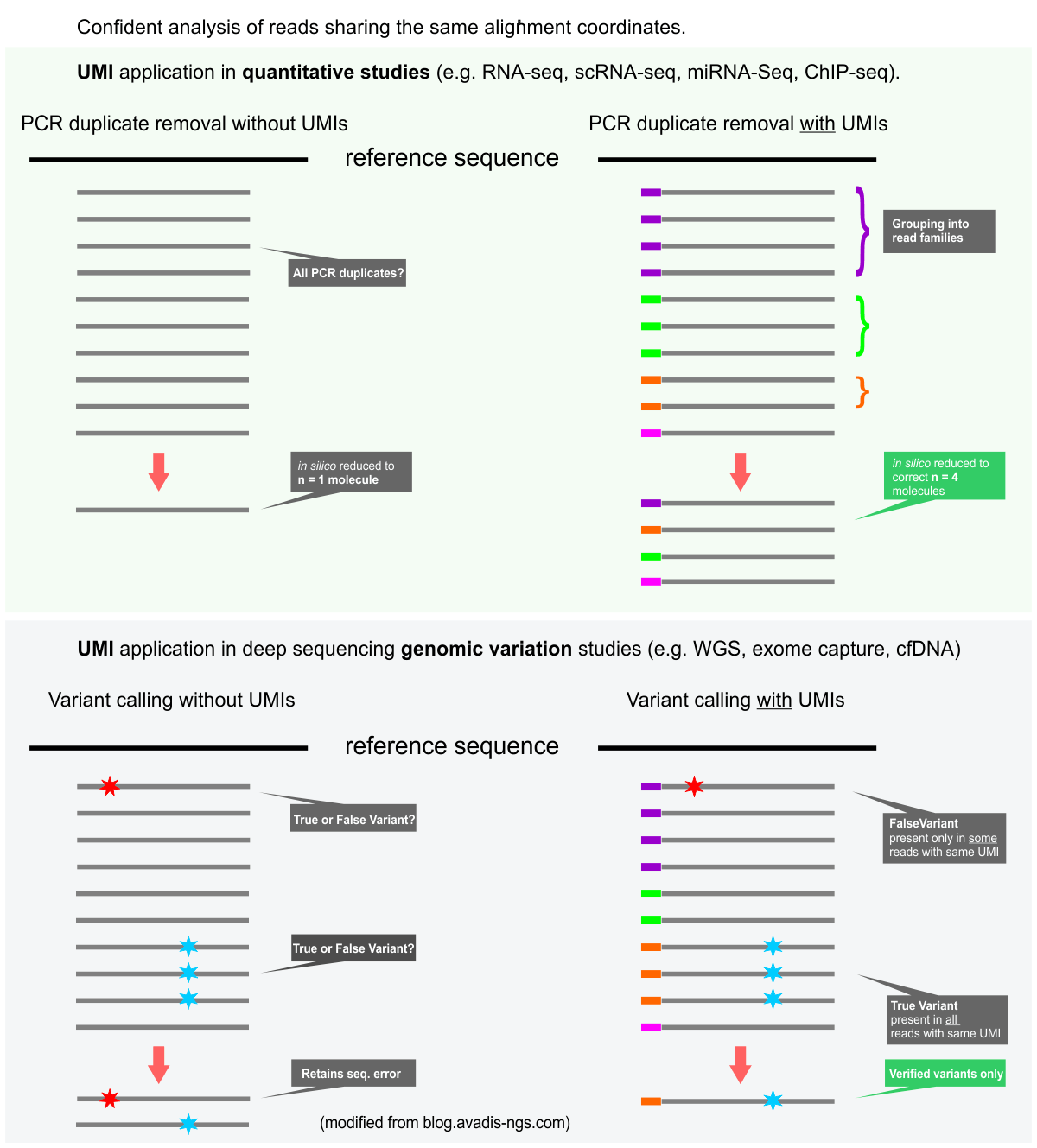
UMIs are valuable tools for both quantitative sequencing applications (e.g. RNA-Seq, ChIP-Seq) and also for genomic variant detection, especially the detection of rare mutations. UMI sequence information in conjunction with alignment coordinates enables grouping of sequencing data into read families representing individual sample DNA or RNA fragments. Please see the graphic below.
The problems UMIs are addressing:
– Quantitative analysis: Many sequencing library preparation protocols enable high-throughput sequencing (HTS) from low amounts of starting material. Their preparation requires PCR amplification of the libraries. While the PCR polymerases and reagents have been improved greatly in recent years enabling a mostly unbiased amplification of sequencing libraries, some biases still remain against sequences with extreme GC contents and against long fragments. When starting from ultra-low input samples, stochastic effects in the first rounds of the PCR add to the problems. These issues can potentially cause erroneous quantitation data. Removal of PCR duplicates using alignment coordinate information is especially inefficient such for low input situations but also for deep sequencing data. In the latter case alignment coordinate-based de-duplification will remove large numbers of biological duplicate reads from the data, especially for the most abundant transcripts.
UMIs alleviate the PCR duplicate problem by adding unique molecular tags to the sequencing library molecules before amplification.
Please also see our FAQ: “Should I remove PCR duplicates from my RNA-seq data?” for more information.
– Rare variant analysis: Illumina sequencing provides data with low error rates (~0.1 to 0.5%) for most applications. These low error rates nevertheless interfere with the confident identification of low abundance variants. UMI-less data can’t distinguish between these and sequencing errors. UMIs in combination with deep sequencing yielding multiple reads for each of the sample DNA fragments solved this problem. The approach was first described as Duplex Sequencing. Hereby, single-strand consensus sequences (SSCSs) and Duplex consensus sequences (DCSs) assembly of the read families increase the accuracy of the sequencing data significantly. Please note that the DNA sample starting amounts and the library yields have to be controlled for this approach to be efficient. Applications include sequencing of heterogeneous tumor samples, cfDNA sequencing including ctDNA sequencing, deep exome sequencing.
The usage of UMIs is recommended primarily for three scenarios: very low input samples, very deep sequencing of RNA-seq libraries (> 80 million reads per sample), and the detection of ultra-low frequency mutations in DNA sequencing. For many other types of projects, UMIs will yield minor increases in the accuracy of the data. In addition, UMI analysis is an excellent QC tool of library complexity.
Incorporating UMIs into sequencing libraries:
– Our 3′-Tag-RNA-Seq protocols employ UMIs by default . For Tag-seq the first 6 bases of the forward read represent the UMI. These are followed by a common linker with the sequence “TATA”, followed by the 12 bp random priming sequence. It is recommended to transfer the UMI sequence information to the read header and to trim the first 22 bases from each read with UMI-TOOLS or custom scripts.
– For conventional RNA-seq and DNA sequencing applications you will specifically have to request UMIs on the submission form. The default library preparations will NOT use UMIs. The UMIs will be located in-line with the insert sequences for conventional RNA-seq, genomic DNA-sequencing, or ChIP-seq. The first twelve bases of both forward and reverse reads will represent UMIs and associated linker sequences (7 nt UMI sequence followed by a 5 nt spacer “TGACT”; UMIs of forward and reverse read are independent resulting in a combined UMI length of 14nt). UMIs and spacer are then followed by the biological insert sequences (for paired-end data a total of 22 bp will be dedicated to the UMIs instead of the inserts). The UMI and spacer sequences are usually trimmed off and the information transferred into the read ID header with software utilities like UMI-Tools or FASTP.
The figure below displays the (simplified) principles of the UMI data analysis for quantitative and variant detection studies.
References:
Parekh et al 2016: The impact of amplification on differential expression analyses by RNA-seq. and
Fu et al. 2018: Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers.
Kennedy et al. 2015: Detecting ultralow-frequency mutations by Duplex Sequencing.
König et al. 2010: iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution.
Smith et al. 2017: UMI-tools: Modelling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy.
Software:
UMI-Tools: https://github.com/CGATOxford/UMI-tools
zUMIs: https://github.com/sdparekh/zUMIs
fastp: https://github.com/OpenGene/fastp (transfer of UMIs into read IDs)
← FAQ