
Summary
UC Davis Genome Center DNA Technologies Core adds three key technologies:
- Nanopore sequencing on the PromethION sequencer
- High Molecular Weight-DNA isolation
- Hi-C Chromatin Conformation Capture
Introduction
The DNA Technologies Core at the Genome Center is adding three services that will be greatly enabling whole genome studies and de novo genome assembly projects. The Genome Center is expanding into the latest sequencing technology, Nanopore sequencing on the PromethION sequencer. This technology can provide the longest sequencing reads, extending to hundreds of thousands of DNA bases. To our knowledge, UC Davis is the first university to PromethION sequencing in an academic core laboratory. The Nanopore sequencing data will enable the assembly of difficult genome regions that so far have escaped analysis. The technology will further significantly reduce the cost of whole genome studies with long reads. The new Nanopore services are complemented by high-molecular weight DNA (HMW-DNA) isolation and Hi-C sequencing library preparation services. The latter technology enables chromosome-scale genome scaffolding. Herewith, UC Davis can now offer all cutting edge technologies enabling genome assemblies. This includes PacBio sequencing, 10X Genomics Chromium linked-read sequencing, PromethION Nanopore and Hi-C sequencing at the DNA Technologies Core as well as optical genome mapping using the Bionano Saphyr in the lab of Mingcheng Luo. The Genome Center Bioinformatics Core is proficient in combining multiple data types into high-quality genome assemblies. Generating highest quality sequencing data for genome assembly projects has already been a focus of the lab for several years. The Core lab has so far contributed long-read (PacBio) and linked-read sequencing to over 240 “large genome” projects. The implementation of the new technologies was supported by a grant of the UC Davis Office Of Research.
The new Technologies
Nanopore sequencing with Oxford Nanopore Technologies (ONT) systems enables high-throughput long-read sequencing of both DNA and RNA samples. For high molecular weight DNA (HMW-DNA) samples read lengths of several hundred kb can be reached with ultra-long-read protocols. The nanopore sequencing data greatly enable de novo genome assemblies and structural genomic variant and transcriptome studies. We offer Nanopore sequencing on the highest throughput Nanopore sequencer, the PromethION. Depending on the samples and the library types, the PromethION, can generate up to 120 Gb of sequence data per flow cell and can currently run up to 24 flow cells simultaneously. The average sequence data yields can vary widely and will mostly depend on the sample properties (please see below). Average yields will be inversely correlated to the library molecule lengths (insert sizes). The best run metrics mentioned below have been achieved in house with DNA isolated from human/mammalian cell cultures and mammalian blood samples.

We offer different types of PromethION sequencing:
- Long-read DNA sequencing: of sheared DNA fragments in the range from 5 kb to 20 kb. Yields per PromethION flowcell can range from 20 Gb up to 120 Gb.
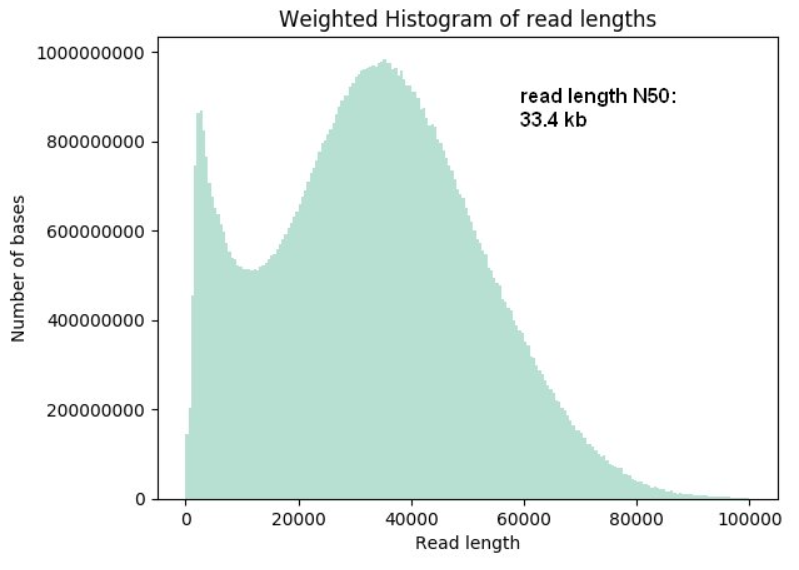
- Super-long-read DNA sequencing: For this protocol we shear DNA to fragments of about 50 kb lengths and are ligating on the sequencing adapters. The yields per flow cell can vary from 20 Gb to up to 90 Gb and read lengths N50 values can reach up to 33 kb and longest reads up to 270 kb length.
- Ultra-long-read DNA sequencing: The protocol is currently being implemented. The sequencing adapters are hereby added by a transposase instead of a ligases. The goal is a significant proportion of reads with lengths longer than 100 kb. The yields are expected to be significantly lower compared to the super-long reads. This type of sequencing is already possible on the MinION sequencer where we have seen read lengths of up to 840 kb and read lengths N50 of up to 70 kb (MinION). It is expected that a combination of super-long-read and ultra-long read data will assist de novo genome assembly projects tremendously.
- Full length cDNA sequencing: The cDNAs will be prepared after priming from the poly-A tails analogous to the Clontech SMART-seq protocol and PCR amplified. The cDNA amplicons will be converted to Nanopore libraries via adapter ligation. These data are used for isoform analysis and gene annotations. In contrast to the PacBio Iso-Seq the protocol the data cannot be processed for high accuracy circular consensus information (CCS) though.
- Direct RNA sequencing: This protocol is being implemented. The library preparation adds an adapter via annealing to the poly-A tail. The sequencing data can be used to investigate RNA-base modifications.
In contrast to other sequencing technologies, the PromethION sequencer is comparatively robust, since it runs only electronics – the chemistry is confined to the disposable flowcells. Thus, UC Davis scientists can get trained to sequence on the PromethION themselves during downtime periods. For collaborative projects we also sequence on the MinION. Oxford Nanopore is expected to offer a flowcell for small-scale test sequencing soon – the so called “Flongle”.
High-Molecular-Weight DNA Isolation: Since high quality single-molecule sequencing data depend on highest quality DNA samples, we now offer HMW-DNA isolation as a service (currently for animal samples). 10X Genomics linked-read sequencing has similarly high sample requirements. The applied DNA isolation protocols will vary with the sample types. Our protocols aim to isolate HMW-DNA samples with mean fragment lengths of 50 kb, or higher, if the sample allows. Longer fragments can be expected for examples for cell culture or fresh blood samples. The HMW-DNA isolation service includes DNA sample QC by pulsed field electrophoresis (PFGE), spectrometry and fluorometry.
Hi-C is a genome-wide Chromatin Conformation Capture protocol using proximity ligation. The technology is of special interest for two areas of research: Studies of the three-dimensional genome organization in the nucleus (e.g. topologically-associating-domains; TADs) as well as chromosome-scale scaffolding of genome assemblies. At the moment we do offer hi-C services for animal samples. We are working on a protocol for plant samples.
Our Nanopore sequencing, HMW-DNA, and Hi-C expert is Ruta Sahasrabudhe, PhD. Please contact her at rmsaha@ucdavis.edu for questions about the new protocols.