
FASTQC is primarily designed to QC whole-genome shotgun sequencing data. Importantly, it is significantly limited in its analyses because it only works on single reads instead of read-pairs.
As a consequence FASTQC tends to generate unnerving warnings for multiple Illumina sequencing data types; This often includes unnecessary warnings about sequence duplication levels and overrepresented sequences.
More modern tools like HTStream and FASTP use more sophisticated algorithms for these purposes and also work on paired-end read data. We highly recommend analyzing duplication levels with these newer tools.
Why does FASTQC show unexpectedly high sequence duplication levels (PCR-duplicates)?
The main reason is that FASTQC only considers single-end sequencing data. All DNA-seq and RNA-seq library preparation protocols involve multiple processing steps. Physical, chemical and enzymatic reactions, all have some sequence-specific biases. As a consequence, the read-start sites are not distributed perfectly random across genomes or transcripts and analyzing a single read start site is insufficient to determine duplicates. Thus, the rate of duplicate reads will always be overestimated and be significantly higher than assumed from read coverage data. Exacerbated is this phenomenon for RNA-seq data which are often dominated by the transcripts of a few genes. For example, it is not uncommon that 50 % of all reads align to the ten most highly expressed genes. The extremely high-read coverage for the particular highly expressed transcripts for RNA-seq data can easily lead to FASTQC read duplication levels of 70% or higher.
Much more realistic read duplication levels can be estimated when incorporating two data points, the read start sites for both forward and reverse reads into the analysis as it is done by HTSream and FASTP.
In addition to only analyzing single reads, FASTQC only analyses 50 nt of the first 100,000 reads for each file for the duplication analysis and extrapolates the dedication rates from this limited number of reads. (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/Help/3%20Analysis%20Modules/8%20Duplicate%20Sequences.html)
Please also see these FAQs:
Should I remove PCR duplicates from my RNA-seq data?
and
What are UMIs and why are they used in high-throughput sequencing?
_____
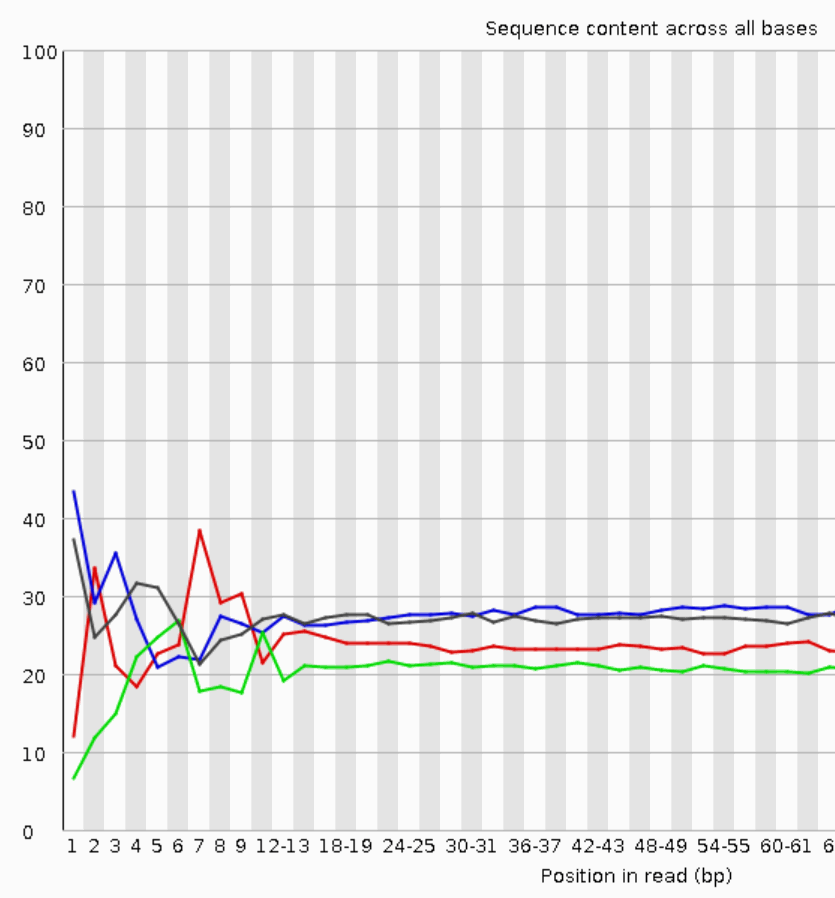
The deviations from the base composition averages at the start of the sequencing data indicate sequence-specific biases of the library preparation chemistry (here Nextera tagmentation data, which show the most obvious bias).
← FAQ