
06 Sequencing Data
By default you will receive gzip compressed FASTQ data, as individual files for each sample (demultiplexed). The demultiplexing is included in the service if you provide us the barcodes sequences on the submission form.
The files will be available for download from our secure SLIMS server.
You will receive only the reads from clusters passing the Illumina quality filter, also called Illumina chastity filter — please see detailed info below. You will find older recommendations on the internet to also analyze reads from clusters that do not pass the chastity filter. These recommendations are outdated. The filtering is very reliable since several years and it is more or less impossible to find any usable data in the reads that have been filtered out.
Otherwise, the data will be complete. By default we do not trim the sequencing data. We would recommend any quality or adapter trimming to be carried out with third-party tools since they provide better results than the Illumina tools and since there are multiple processing options. SRA submissions also require full-length data.
Please note that the sequence data can contain traces of the Illumina PhiX internal standard. For applications like genome assemblies, these PhiX reads should be removed. BBduk is a free software to achieve this. Please see the Kmer-filtering paragraph in the BBduk help. Please also see the preprocessing section in this presentation: https://ucdavis-bioinformatics-training.github.io/2017-June-RNA-Seq-Workshop/tuesday/Preprocessing.pdf
The Illumina Chastity Filter:
The Illumina chastity filter is applied only to the first 25 bases of the forward read data per cluster. The fluorescence intensity ratios are calculated; specifically the chastity value is defined as the ratio of the brightest base intensity divided by the sum of the brightest and second brightest base intensities. Clusters pass the filter if no more than 1 base call has a chastity value below 0.6 in the first 25 cycles.
Please see this FAQ:
When should I trim my Illumina reads and how should I do it?
Should I trim adapters from my Illumina reads?
This depends on the objective of your experiments. For counting applications such as differential gene expression (DGE), RNA-seq analysis, ChIP-seq, or ATAC-seq, read trimming is generally not required anymore when using modern aligners. For such studies local aligners or pseudo-aligners should be used. Modern “local aligners” like STAR, BWA-MEM, HISAT2, will “soft-clip” non-matching sequences. Pseudo-aligners like Kallisto or Salmon will also not have any problem with reads containing adapter sequences.
However, if the data are used for variant analyses, genome annotation or genome or transcriptome assembly purposes, we recommend read trimming, including both adapter and quality trimming.
DNA and RNA sequencing:
Truseq forward read: AGATCGGAAGAGCACACGTCTGAACTCCAGTCA
Truseq reverse read: AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT
DNA sequencing:
Nextera: CTGTCTCTTATACACATCT
For small RNA/miRNA sequencing data please use this sequence, but also see this FAQ: How should the miRNA/smallRNA data be trimmed?.
TruSeq Small RNA: TGGAATTCTCGGGTGCCAAGG
Should I remove PCR duplicates from my RNA-seq data?
The short and generalized answer to the question “Should I remove PCR duplicates from my RNA-seq data?” is in most cases NO. For some scenarios, de-duplification can be helpful, but only when using UMIs. Please see the details below.
The vast majority of RNA-seq data are analyzed without duplicate removal. Duplicate removal is not possible for single-read data (without UMIs). De-duplification is more likely to cause harm to the analysis than to provide benefits even for paired-end data (Parekh et al. 2016; below). This is because the use of simple sequence comparisons or the typical use of alignment coordinates to identify “duplicated reads” will lead to the removal of valid biological duplicates. RNA-seq library preparation involves several processing steps (e.g. fragmentation, random priming, A-tailing, ligation); none of these processes is truly random or unbiased. Thus, the occurrence of “duplicated reads” in between millions of reads can be expected even in paired-end read data. Short transcripts and very highly expressed transcripts will show the majority of such “natural” duplicates. Their removal would distort the data. For example plant RNA-seq data often seem to contain large amounts of duplicated reads. This is in part due to the fact the gene expression in many plant tissues, like leaves, is dominated by a small number of transcripts; much more so than in most animal samples. Another concern is that the fraction of reads identified as “duplicated” is correlated to the number of aligned reads. Thus, one would have to normalize any data set for equal read numbers to avoid introducing additional bias.
Several studies (among them Parekh et al. 2016; below) have shown that retaining PCR- and Illumina clustering duplicates does not cause significant artifacts as long as the library complexity is sufficient. The library complexity is in most cases directly related to the amount of starting material available for the library preparation. Chemical inhibitors present in the sample could also cause low conversion efficiency and thus reduced library complexities.
PCR duplicates are thus mostly a problem for very low input or for extremely deep RNA -sequencing projects. In these cases, UMIs (Unique Molecular Identifiers) should be used to prevent the removal of natural duplicates. UMIs are for example standard in almost all single-cell RNA-seq protocols.
The usage of UMIs is recommended primarily for two scenarios: very low input samples and very deep sequencing of RNA-seq libraries (> 80 million reads per sample). UMIs are also employed for the detection of ultra-low frequency mutations in DNA sequencing (e.g. Duplex-Seq). For other types of projects, UMIs will have a minor effect in reducing PCR amplification induced technical noise.
Our 3′-Tag-RNA-Seq protocol employs UMIs by default. For other RNA-seq applications please request UMIs on the submission form. When using UMIs for conventional RNA-seq, genomic DNA-sequencing, or ChIOP-seq, the first eleven bases of both forward and reverse reads will represent UMI and linker sequences. These are then followed by the biological insert sequences. The UMI sequences are usually trimmed off and the information transferred into the read ID header with software utilities like UMI-Tools.
Please see the discussion here for details:
https://www.biostars.org/p/55648/ and these excellent papers
Parekh et al 2016: The impact of amplification on differential expression analyses by RNA-seq. and
Fu et al. 2018: Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers.
Kennedy et al 2015: Detecting ultralow-frequency mutations by Duplex Sequencing.
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
This blog post “molecularecologist.com/2016/08/the-trouble-with-pcr-duplicates” offers a detailed analysis of the effect of increasing read numbers on the frequency of PCR duplicates as well as the occurrence of false-positive duplicate identifications on another type of Illumina sequencing data (RAD-seq). Please note that the library type studied is different from RNA-seq as are the potential effects of PCR duplicates for this type of analysis. In contrast to RNA-seq, PCR duplicates should be removed for most RAD-seq studies.
FASTQC is primarily designed to QC whole-genome shotgun sequencing data. Importantly, it is significantly limited in its analyses because it only works on single reads instead of read-pairs.
As a consequence FASTQC tends to generate unnerving warnings for multiple Illumina sequencing data types; This often includes unnecessary warnings about sequence duplication levels and overrepresented sequences.
More modern tools like HTStream and FASTP use more sophisticated algorithms for these purposes and also work on paired-end read data. We highly recommend analyzing duplication levels with these newer tools.
Why does FASTQC show unexpectedly high sequence duplication levels (PCR-duplicates)?
The main reason is that FASTQC only considers single-end sequencing data. All DNA-seq and RNA-seq library preparation protocols involve multiple processing steps. Physical, chemical and enzymatic reactions, all have some sequence-specific biases. As a consequence, the read-start sites are not distributed perfectly random across genomes or transcripts and analyzing a single read start site is insufficient to determine duplicates. Thus, the rate of duplicate reads will always be overestimated and be significantly higher than assumed from read coverage data. Exacerbated is this phenomenon for RNA-seq data which are often dominated by the transcripts of a few genes. For example, it is not uncommon that 50 % of all reads align to the ten most highly expressed genes. The extremely high-read coverage for the particular highly expressed transcripts for RNA-seq data can easily lead to FASTQC read duplication levels of 70% or higher.
Much more realistic read duplication levels can be estimated when incorporating two data points, the read start sites for both forward and reverse reads into the analysis as it is done by HTSream and FASTP.
In addition to only analyzing single reads, FASTQC only analyses 50 nt of the first 100,000 reads for each file for the duplication analysis and extrapolates the dedication rates from this limited number of reads. (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/Help/3%20Analysis%20Modules/8%20Duplicate%20Sequences.html)
Please also see these FAQs:
Should I remove PCR duplicates from my RNA-seq data?
and
What are UMIs and why are they used in high-throughput sequencing?
_____
The deviations from the base composition averages at the start of the sequencing data indicate sequence-specific biases of the library preparation chemistry (here Nextera tagmentation data, which show the most obvious bias).
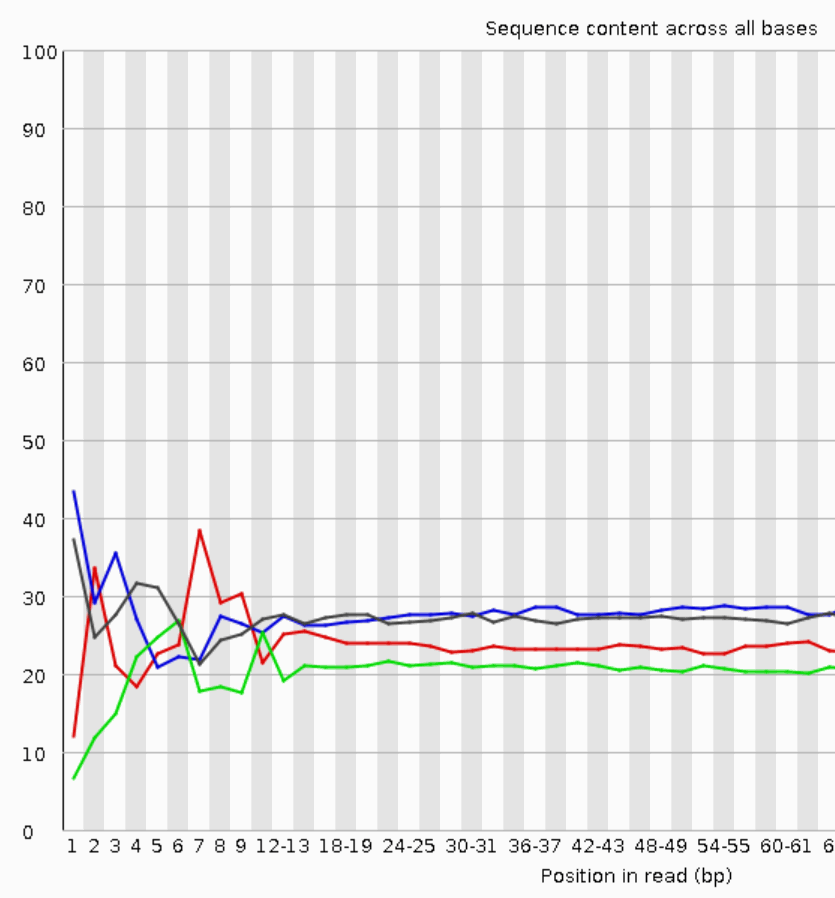
Please note that the UMIs provide optional additional data analysis options; for many applications, the UMI information can be safely ignored. UMIs are especially beneficial for low RNA input situations as well as ultra-deep sequencing.
For push-button type secondary analyses (combining data for up to 2 SMRT-cells e.g. for demultiplexing, CCS, long amplicon, or IsoSeq analysis) we can run these on our own server and will also deliver all the resulting data. Please note that we are not data analysts, and we can not experiment with analysis parameters. Thus, we highly recommend to work with the Bioinformatics Core for comprehensive data analyses. Alternatively it is recommended that you should install the SMRT Tools command line programs which are part of the SMRT-Link package.
The data files generated by the PacBio Sequel are different from the data generated by the PacBio RSII previously. All the data that were before contained in the bas.h5/bax.h5 and accessory formats are now contained in the .bam, .xml, and .pbi files. Please see this page for the detailed format specifications (pacbiofileformats.readthedocs.io/en/5.0/index.html). Specifically the raw data or each SMRT-cell will be in files named .subreads.bam, .subreads.xml, and .subreads.pbi .
The .bam data can be converted to fastq or fasta files with bamtools (please see at the bottom of this page: github.com/PacificBiosciences/PacBioFileFormats/wiki/BAM-recipes) or best with the PacBio tool bam2fastx. “bam2fastx” is part of the free SMRT Tools: pacb.com/support/software-downloads/ .
!). PacBio came to the conclusion that computing the quality scores for the raw data was a waste of time. Apparently the quality scores for the raw data cannot be reliably computed (and consequently these were also ignored for RSII data pipelines). However, usable PacBio quality scores can be generated from consensus data if the project allows (either by CCS or other secondary analysis algorithms: e.g. by alignments all-vs-all). In short the determination of the quality of individual reads is up the downstream analysis pipeline (e.g. the assembler).We deliver sequencing data via two portals: SLIMS for Illumina data, and BioShare for PacBio and Nanopore data. Both portals offer secure access to the data and support several download protocols. The emails that will notify you about new sequencing data on SLIMS will contain download instructions.
The SLIMS download instructions are available here: https://dnatech.genomecenter.ucdavis.edu/archiving-slims-data/
In BioShare the instructions for several download protocols are integrated into the user interface – BioShare will automatically generate the download commands for you.
Since high throughput sequencing data files tend to be big, we recommend downloading the data by running rsync or wget command line tools (built-in on Linux/Mac/Cygwin). Wget can be easily added to Windows10 as part of a full-featured Linux subsystem (including bash, rsync, wget, …) . This Linux can run in parallel with the Windows GUI. Please see instructions here: How to install WSL on Windows 10 .
The easiest way to add command line UNIX/Linux/BASH functionality (including rsync and wget) to other Windows versions is to install the free MobaXterm terminal from here: https://mobaxterm.mobatek.net/
Please note: We do NOT archive sequencing data generated for you. Any sequencing data should be downloaded and verified as soon as possible.
Illumina sequencing data will be available for download for three months after they are generated. Pacbio and Nanopore data will be available for two months.
Be default we will demultiplex all sequencing data from libraries generated by our facility as well as from customer libraries with barcodes sequenced in separate indexing reads (e.g. using Truseq-style adapters). This is in contrast to adapters with old style in-line barcode data. Data for these have to be demultiplexed by the customer.
The de-multiplexing is included in the sequencing recharge rates. There are no additional costs.
Following analysis of each run, users have access to parsed output through the SLIMS server. A SLIMS account will be created for you on your first run, with information about how to access your account distributed via email (you will receive this email before your actual files are available). The main SLIMS page can be reached here.
You can download all your files from SLIMS with your webbrowser (clicking the links) or better with a download manager (e.g for Firefox https://addons.mozilla.org/en-US/firefox/addon/downthemall/ )
However, it is recommended to download with command line tools. E.g. by running “wget” (built-in into Unix/Linux/Mac/Cygwin operating systems, available also for Windows*) with a command like the following:
wget -r -nH -nc -np -R index.html* “http://slims.bioinformatics.ucdavis.edu/Data/Your_RANDOM_STRING/” &
This command will download all files into your current working directory.
You can also archive all your data with RSYNC by following instructions at
http://wiki.bioinformatics.ucdavis.edu/index.php/Archiving_solexa_data .
*The easiest way to get rsync and wget tools on Windows is by installing the free MobaXterm terminal.
Please note: We do NOT archive sequencing data generated for you. Any sequencing data should be downloaded and verified as soon as possible.
Illumina sequencing data will be available for download for three months after they are generated. Pacbio and Nanopore data will be available for two months.
We do NOT archive sequencing data generated for you. Any sequencing data should be downloaded and verified as soon as possible.
The sequencing data will be available to you on our SLIMS server (Illumina data) or Bioshare server (PacBio and Nanopore data). Illumina sequencing data will be available for download for three months after they are generated. Pacbio and Nanopore data will be available for two months.
You will receive emails from SLIMS or Bioshare notifying you as soon as the data are available for download.
It is highly recommended to download and verify your data (especially the demultiplexing results) at your earliest convenience after receiving the email notification.
We are using the PerkinElmer NEXTflex™ Small RNA-Seq kit for the generation of micro RNA and small RNA-seq libraries because it significantly reduces sequence-specific biases in the library preparation. For this purpose the adapters oligonucleotides contain 4 randomized bases at the ligation junctions. These randomized bases should be removed by trimming before mapping the sequence reads. BiooScientific recommends this procedure:
Data Analysis for micro RNA data generated with the PerkinElmer kits:
The 3′ and 5′ adapters included in this kit both contain 4 random bases that will appear immediately 5′ and 3′ to the insert in sequencing data. The presence of these random bases should be considered when choosing an alignment strategy. When using “end-to-end” alignment, we recommend processing data in the following manner: 1. Clip the 3′ adapter sequence (TGGAATTCTCGGGTGCCAAGG ; this sequence fragment is sufficient!). 2. Trim the first and last 4 bases from the adapter-clipped reads. 3. Perform alignments as normal. Alternatively, alignment may be performed in “local” mode.
Please find the full adapter sequences for your reference here: Bioo-Scientific-Small-RNA-Barcode-Indices-v1-1-15 adapters barcodes small RNA micro-RNA miRNA
Strand-Specific RNA-Seq Libraries
RNA-Seq (conventional) after Poly-A enrichment or ribodepletion:
By default we generate strand-specific RNA-seq libraries. Strand-specific (also known as stranded or directional) RNA-seq libraries substantially enhance the value of an RNA-seq experiment. They add information on the originating strand and thus can precisely delineate the boundaries of transcripts in regions with genes on opposite strands.
There are several ways to accomplish strand-specificity. We incorporate dUTP during the second-strand synthesis of the cDNA. The dUTP containing strand will not be amplified by the proofreading polymerase used for library amplification, thus preserving the strand information for RNA-seq.
For single-end sequencing, the resulting data will represent the “anti-sense strand”. When using paired-end sequencing, the forward read of the resulting sequencing data represents the “anti-sense strand” and the reverse read the “sense strand” of the genes (for Trinity transcriptome assemblies the “–RF” orientation flag should be used). Illumina paired-end reads are always inward oriented (with the exception of “jumping” or “mate-pair” libraries).
Tag-Seq:
Tag-seq data are strand-specific and have a “sense-strand” orientation.
small-RNA-Seq/miRNA-seq:
Small RNA-seq data are strand-specific. The forward read of the sequencing data (read 1) is oriented as the reverse complement of the original RNA molecule. For libraries generated with the Revvity (PerkinElmer) Nextflex small RNA kits, the RNA sequence will be both preceded and followed by four bases with a random sequence.