New High-Throughput Gene-Expression Analysis Services (3’Tag-Seq)
The DNA Technologies and the Bioinformatics Cores have been working for more than half a year with a simplified RNA-Seq protocol that promises to drastically reduce the costs of many gene-expression studies, while fully maintaining the precision of the analysis.
The new approach is called 3’Tag-Seq (or TagSeq, 3’Tag-RNA-Seq, Digital RNA-seq but these names have also been used for a variety of different protocols). In contrast to traditional RNA-Seq, which generates sequencing libraries from the whole transcripts, 3-Tag-Seq only generates a single initial library molecule per transcript, complementary to 3′-end sequences. For example for human samples, the restriction to a small part of the transcripts reduces the number of sequencing reads required by at least five times. In contrast to earlier “digital RNA-seq” protocols that were based on restriction digestions of cDNAs, the current protocol combines reverse transcription priming from the poly-A tail with random priming and adapter placement for the second-strand synthesis. In most cases up to 48 samples can be sequenced per HiSeq 4000 lane.
More than 90% of the RNA-seq studies carried out in our labs are analyzed exclusively for differential gene expression (DGE). The conventional full transcript RNA-seq protocols generate more data than needed for this specific purpose, but they also allow for splicing analyses. The complexity of the standard RNA-seq data is not an advantage if the aim of the project is only DGE analysis – 3’Tag-Seq might actually be the superior tool for this application (DGE). In our experience the 3’Tag-Seq data have so far shown exceptionally low noise as well as insensitivity to RNA sample quality variations.
This example MDS plot shows an analysis of 3’Tag-Seq data of macrophage cells exposed to three types of bacterial infections and mock-infections at two time points. The analysis distinguishes the responses to the individual bacterial species and the duration of the infections. Even the reactions to the mock-infections are clustered by time points.

We are currently offering 3’Tag-Seq as a low cost custom sequencing service but are planning to offer 3’Tag-Seq services soon at simple per-sample recharge rates — including both library preps and sequencing. In the long run the services can also include a basic differential-gene-expression analysis.
Advantages of 3’Tag-Seq:
- low noise gene expression profiling
- less sensitive to RNA sample quality/integrity variations (compared to poly-A enrichment protocols)
- requires significantly lower numbers of sequencing reads
- single read sequencing is sufficient
- simpler library prep protocol
- costs about half or less compared to standard RNA-seq
- costs lower than, or comparable to, microarray analysis
- much higher dynamic range compared to microarrays
- soon: simple pricing scheme and simplified planning of experiments
Disdavantages of 3’Tag-Seq:
- data do not contain any transcript-splicing information
- data analysis requires a reference genome with good annotation (also of the UTRs)
- only applicable to eukaryotic samples
- protocol is a (a bit) more sensitive to chemical contaminants (spin column cleaned RNA samples are recommended)
Index Mis-Assignments on HiSeq 4000 and HiSeq X
Discussions with Illumina have confirmed that there exists a problem of low-level index mis-assignments for all sequencers using the Exclusion-Amplification clustering chemistry (HiSeq 3000, 4000, and X). For most projects this does not constitute a problem due to the low rate – however it could affect the analyses of low-frequency variants. We will use a set of uniquely dual-indexed adapters for such projects to be able to filter out such mis-assignments.
For more information please also see: http://enseqlopedia.com/2016/
High-Throughput Sequencing on the Illumina NextSeq
In collaboration with the Foundation Plant Services we can now offer high throughput sequencing on the Illumina NextSeq system. The NextSeq reagents come in 75-cycle, 150-cycle, and 300-cycle capacities with high-output and mid-output flowcells (150 and 300 cycles only). The official specs are up to 400 million and and up to 130 million clusters passing filter, respectively, for these flowcells. However, with optimal sequencing libraries significantly higher read number can be achieved.

The advantages of the NextSeq are:
- very fast turnaround times
- allows custom-sequencing primers
- most affordable high-throughput paired-end
sequencing data (for paired-end 40 bp reads) - for optimal shorter-insert sequencing libraries the numbers of clusters passing filter can reach 600 million per run
- allows asymmetrical read lengths
Drawbacks of the Nextseq are:
- high-output 150 and 300-cycle kits are comparatively pricey
- the sequence uses a two-color chemistry; thus not necessarily suited for de-novo assembly projects; however there is no concern for all “counting applications” (e.g. RNA-seq, ChIP-seq).
High-Throughput Library and Sample Size-Selection on the Pippin HT
Our Core has acquired a Pippin HT sample sizing instrument that allows to more precisely size-select DNA samples and sequencing libraries. Twenty-four samples can be processed on a single run.

The instrument belongs to our Shared Equipment – this means all UCD researchers can be trained to use this instrument.
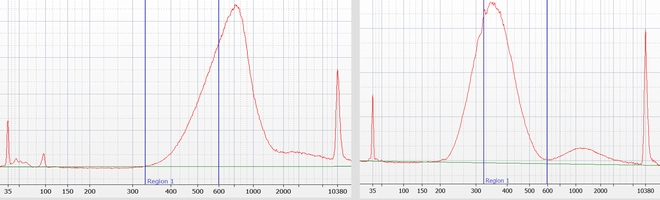
We will use this precise size-selection for all genomic DNA sequencing libraries prepared by our lab. This allows us to avoid generating redundant data for short insert library molecules (for which the reads would otherwise overlap); the precise size selection will further avoid lower quality data caused by library molecules that are too long (e.g. longer than 600 bp for the HiSeq 4000). For the two example customer libraries size selection for the optimal sizes (within the blue bars) removed about 50% of the library molecules that are outside of the optimal range.