
04 Library Preparation and QC
Ampure XP bead “upper cut” protocol to remove fragments longer than 670 bases:
-
-
- If not mentioned explicitly follow the standard Ampure XP handling instructions from the manufacturer (e.g. equilibrate the beads at to room temperature before use; vortex beads before use, details of the bead washes and elution,…)
- If the sample volume is smaller than 50 ul, add EB buffer up to 50 ul to each sample.
- Add 0.55x the sample volume in Ampure beads (e.g. 27.5 ul beads to a 50 ul sample) to your sample, mix, incubate for 5 minutes at RT.
- Collect the beads on a magnet.
- Transfer the supernatant to a new tube.
- Add another 1x original volume Ampure beads to the supernatant; mix; incubate for 5 minutes
- Collect the beads on a magnet and remove the supernatant
- Carry out the two regular 80% ethanol washes of the beads and elute the samples from the beads according to Agencourt Ampure XP protocol.
- Verify the success of the size selection by running an aliquot on a Bioanalyzer or equivalent instrument.
-
Ampure XP bead “upper & lower cut” protocol to remove fragments longer than 670 bases and shorter than 400 bases:
This protocol is identical to the one above but adds a smaller volume of the beads at step 6 for the final enrichment onto the beads. The reduced bead buffer concentration at this step leads to a removal of longer fragments compared to the protocol above.
Please note: It is recommended to verify this protocol first with your batch of Ampure XP beads or similar beads from other manufacturers. Bead-based size selection cannot carry out precise “cuts”; thus, you will also lose some of the library in the size ranges that you intend to keep.
- If not mentioned explicitly follow the standard Ampure XP handling instructions from the manufacturer (e.g. equilibrate the beads at to room temperature before use; vortex beads before use, details of the bead washes and elution,…)
- If the sample volume is smaller than 50 ul, add EB buffer up to 50 ul to each sample.
- Add 0.55x the sample volume in Ampure beads (e.g. 27.5 ul beads to a 50 ul sample) to your sample, mix, incubate for 5 minutes at RT.
- Collect the beads on a magnet.
- Transfer the supernatant to a new tube.
- Add another 0.25x of the original volume Ampure beads (e.g. 12.5 ul beads for a sample of a 50 ul starting volume sample) to the supernatant; mix; incubate for 5 minutes
- Collect the beads on a magnet and remove the supernatant
- Carry out the two regular 80% ethanol washes of the beads and elute the samples from the beads according to Agencourt Ampure XP protocol.
- Verify the success of the size selection by running an aliquot on a Bioanalyzer or equivalent instrument.
Beckmann/Agencourt also sells beads that are dedicated to size selections named SPRIselect — however, very likely these are actually identical to the AMpure XP beads. The SPRIselect manual provides a lot of additional information and protocols that can be applied to AMpure XP and other beads. Please see here: Beckman SPRIselect Ampure beads
BTW, our favorite magnetic separator for 96-well plates is this one from EdgeBio.
RNA-seq experiments should best be carried out with samples of consistent RNA integrity and input amounts. However, some RNA-seq samples can be so limited and irreplaceable that experiments have to be carried out with less than the recommended input amounts. Similar complications can occur if some of the samples are significantly more degraded than others. Such situations require weighing the pros and cons when choosing the input amounts from the more abundant samples.
Points to consider are:
- The ideal approach for an RNA-seq project would be to treat each sample exactly the same, to minimize technically induced variation in the resulting data. This would include starting each library prep with the same amount of total RNA input and applying the same number of PCR cycles to each of the libraries. However, more degraded samples usually require increased input amounts.
- In general, sequencing library preparations do not fail at a specific input amount threshold. Lower amounts can usually be compensated for by increasing the number of PCR cycles during the preparation. Thus, inputs lower than the kit manufacturers’ recommendations can be used in some cases. Any reduced input amounts (and/or higher sample degradation) will, however, lead to reduced library complexities, and thus noisier gene expression data. The best data are usually generated when working with input amounts in the upper half of the manufacturers’ input recommendations.
For sample sets with varying RNA sample amounts and qualities, we suggest verifying first if outlier samples with significantly lower sample amounts or lower quality can be dropped from the experiment. If this is not the case we suggest two options. We will ask you to pick one of these or to provide detailed instructions for another approach:
Strategy #1: Normalize all RNA input amounts to the lowest mass sample that has to be included in the study. Please note that this will more severely impact the quality for the originally high RNA quality & high RNA amount samples.
Strategy #2: Normalize the RNA input amounts to a range from the lowest input sample to three times that of the lowest input sample. With this approach, all libraries will still undergo the same number of PCR cycles, which preserves more of the sample quality of the more abundant and higher quality samples. (An example case would be that the lowest available amount for one of the samples is 10 ng. We would then dilute only high-amount samples to an input of at most 30 ng.)
For most projects, we tend to recommend Strategy #2, especially if the ratio of the low-input outlier samples is low.
Is PCR-free library preparation still advantageous?
In general, the original concerns about library PCR amplification (presented in papers from 2008) are no longer very relevant. This is due to the use of modern polymerases that are designed for complex samples like Kapa HiFi, NEB Q5, or QIAseq HiFi polymerase. The previous “standard”, the high-fidelity Phusion enzyme had tremendous disadvantages for complex samples (Quail et al. 2012 Optimal enzymes for amplifying sequencing libraries. Nature Methods volume 9, pages10–11(2012) https://www.nature.com/articles/nmeth.1814 ).
PCR-free libraries also have disadvantages, since they require significantly higher library QC efforts. Thus, we are charging a PCR-free Add-On fee for the preparation of PCR-free libraries.
What are your recommendations?
A great alternative to preparing the libraries completely PCR-free is the use of a single PCR cycle instead. This combines the advantages of both: It creates fully double-stranded library molecules that do not cause any problems in the library QC. In addition there will be no or only an extremely low PCR-bias introduced. Our recommendation is to submit the same amount of DNA sample as for PCR-free library preps (e.g. 1 ug) and then request the single PCR cycle library amplification.
Quality and quantity of DNA and RNA is critical for high quality sequencing output. Please make sure your DNA is not degraded and is free of RNA contamination. RNA samples should always be assessed on the bioanalyzer for the absence of gDNA contamination (can be removed with DNaseI treatment followed by a column clean-up; e.g. Zymo “RNA Clean and Concentrator”) and degradation. Preferentially determine the concentrations of your DNA and RNA samples using fluorometry (e.g. with a Qubit or plate reader). The sample purity should be assessed by spectrophotometry (e.g. Nanodrop). Please see this page for a comprehensive table of sample requirements for sample QC, library preps, or your self-made libraries. Please see the Library Prep Page for details on the library prep processes. For submission information, including submission forms and shipping details, please visit the Sample Submission & Scheduling page. If you are submitting DNA for PacBio libraries, please follow the PacBio Guidelines for Shipping and Handling.
The Real-time PCR core can carry out DNA as well as RNA extractions for you.
When designing RNA-seq or ChIP-seq experiments, it is very important to avoid technical replicates and pseudo-biological replicates as they will lead to spurious results (e.g. spurious differential gene expression data; DGE data in case of RNA-seq).
Creating pseudo-biological replicates occurs frequently, especially for in vitro studies. Doing so can often lead to hundreds of false positive differentially expressed genes. For example, treating three cell-culture flasks of the same passage of a cell line as biological replicates would create such a dilemma. Please see the excellent discussion of this topic by Christoph Emmerich here: https://paasp.net/accurate-design-of-in-vitro-experiments-why-does-it-matter/ .
This video by Josh Starmer explains why technical replicates are not helpful in principle in RNA-seq: https://www.youtube.com/watch?v=gKnfP2_Xdpo .
Ampure XP/SPRI bead “upper cut” protocol to remove double-stranded DNA fragments over 670 bases:
- Bead-based size selection cannot carry out precise “cuts”; Thus, you will also lose some of the library molecules in the size ranges that you intend to keep. This selection protocol will also reduce adapter dimers and other molecules shorter than 160 bp.
- It is recommended to verify this protocol first with your batch of beads.
- Multiple other manufacturers offer copies of the Ampure XP product (e.g. SPRI beads, Kapapure, …). These can work just as efficiently. Please test them beforehand.
- The cutoff fragment length can be modified by changing the ratios of SPRI-beads to sample volume.
-
-
- If not mentioned explicitly follow the standard Ampure XP handling extractions from the manufacturer (e.g. equilibrate the beads to room temperature before use; vortex beads before use, details of the bead washes and elution,…)
- If the sample volume is smaller than 50 microliters, add molecular biology grade water up to 50 microliters.
- Add 0.55x the sample volume in Ampure XP beads to your sample, mix, incubate for 5 minutes at RT.
- Collect the beads on a magnet.
- Transfer the supernatant to a new tube.
- Add another 1x original volume Ampure beads to the supernatant; mix; incubate for 5 minutes
- Collect the beads on a magnet and remove the supernatant
- Carry out the regular 80% ethanol washes of the beads and elute the samples from the beads according to Agencourt Ampure XP protocol.
- Verify the success of the size selection by running an aliquot on a Bioanalyzer or equivalent instrument.
-
In case the library preparation did not generate sufficient library material required to load a sequencer, Illumina libraries can be amplified with a universal PCR protocol.
While the amplification can rescue experiments, it is worth considering on a per-project basis if perhaps the library preparation should be repeated instead. For quantitative experiments, it is generally recommended to treat all libraries the same throughout the pipeline. Insufficient library yields in the initial library preparation could be signs of sample contamination, processing errors, etc. with potential side effects that cannot be remedied by amplification.
Illumina library amplification PCR protocol:
The library amplification uses standard Illumina P5 (5′-AATGATACGGCGACCACCGAGATCT-3′) and P7 (5′-CAAGCAGAAGACGGCATACGAGAT-3′) PCR primers which can be ordered as desalted DNA oligos.
Create a 10x concentrated primer mix at 10 μM each of these primers in EB buffer.
Add up to 20μl library, add water up to a volume of 20μl, add 5 μl 10x primer mix. Then add 25μl Kapa HiFi 2x Hotstart PCR master mix and pipette up and down several times.
Use the following cycling parameters:
Initial denaturation: 98°C 45 sec,
X PCR amplification cycles consisting of: denaturation 98°C 15 sec, annealing 60°C 30 sec, extension 72°C 30 sec
Final extension 1 min.
Assuming one wants to generate at least 100 ng of sequencing library, we recommend performing four cycles of PCR when starting from 10 ng library.
After the PCR perform a standard Ampure XP/SPRI bead cleanup (e.g. with beads at 1.2x the sample volume, Ampure beads or equivalent) and elute in 30μl EB buffer.
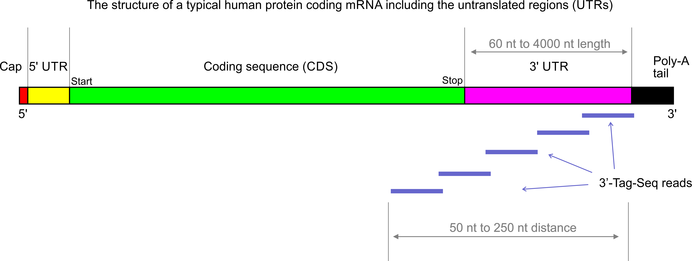
3’Tag-Seq is a protocol to generate low-cost and low-noise gene expression profiling data. The protocol is also known as TagSeq, 3’Tag RNA-Seq, Digital RNA-seq, Quant-Seq (please note that most of these names have also been used for a variety of other protocols previously). In contrast to traditional RNA-Seq, which generates sequencing libraries from the whole transcripts, 3-Tag-Seq only generates a single initial library molecule per transcript, complementary to 3′-end sequences. For example for human samples, the restriction to a small part of the transcripts reduces the number of sequencing reads required by at least five times. In contrast to earlier “digital RNA-seq” protocols that were based on restriction digestions of cDNAs, the current protocol combines reverse transcription priming from the poly-A tail with random priming and adapter placement for the second-strand synthesis. In most cases up to 48 samples can be sequenced per HiSeq 4000 lane.
More than 90% of the RNA-seq studies carried out in our labs are analyzed exclusively for differential gene expression (DGE). The conventional full transcript RNA-seq protocols generate more data than needed for this specific purpose, but they also allow for splicing analyses. The complexity of the standard RNA-seq data is not an advantage if the aim of the project is only DGE analysis – 3’Tag-Seq might actually be the superior tool for this application (DGE). In our experience the 3’Tag-Seq data have so far shown exceptionally low noise as well as insensitivity to RNA sample quality variations.
This example MDS plot shows an analysis of 3’Tag-Seq data of macrophage cells exposed to three types of bacterial infections and mock-infections at two time points. The analysis distinguishes the responses to the individual bacterial species and the duration of the infections. Even the reactions to the mock-infections are clustered by time points.

We are currently offering 3’Tag-Seq as a low cost custom sequencing service but are planning to offer 3’Tag-Seq services soon at simple per-sample recharge rates — including both library preps and sequencing. In the long run the services can also include a basic differential-gene-expression analysis.
Advantages of 3’Tag-Seq:
- low noise gene expression profiling
- less sensitive to RNA sample quality/integrity variations (compared to poly-A enrichment protocols)
- >99% strand-specific; same direction as mRNA transcripts
- requires significantly lower numbers of sequencing reads
- single read sequencing is sufficient
- simpler library prep protocol
- costs about half or less compared to standard RNA-seq
- costs lower than, or comparable to, microarray analysis
- much higher dynamic range compared to microarrays
- we routinely sequence 48 libraries per HiSeq lane; for soBarclays
- for very low input or high depth sequencing of 3’Tag-Seq libraries UMI‘s (unique modular identifiers) can be incorporated
- Batch-Tag-Seq packages: simple pricing scheme and simplified planning of experiments
Disdavantages of 3’Tag-Seq:
- data analysis requires a reference genome with good annotation (including UTRs)
- only applicable to eukaryotic samples
- data do not contain any transcript-splicing information
- protocol is (a bit) more sensitive to chemical contaminants (spin column cleaned RNA samples are recommended)
For high-throughput 3’Tag-Seq library generation we require pure total RNA samples at a concentration of 100 ng/ul (best submit 10 ul at 100 ng/ul). For custom 3’-Tag-Seq library preps the input amounts can be a low as 10 ng total. The RNA samples for this protocol need to be isolated or cleaned-up by spin-column protocols. Please also see the sample requirements page.
3′-Tag-Seq libraries are sequenced by single-end sequencing on the HiSeq 4000 or the NextSeq.
Please note that 3’Tag-Seq libraries generate lower read numbers on the HiSeq 4000 (about 320 million reads per lane) compared to standard RNA-seq libraries. Since the DGE analysis of tag-Seq data requires much lower read numbers this is usually not a problem.
The libraries will be sequenced on Illumina HiSeq 4000 or NextSeq 500 sequencers with single-end 80 or 90 bp reads (SE80 or SE90). Please note that for some analysis pipelines it is recommended to trim off the first 12 bases from the reads. We will provide the full length data. Trimming is not necessary if you are using a local aligner (like STAR or BBmap). The sequences can be trimmed easily, for example with the “reformat” command from BBTools. In case UMIs are incorporated, the first 6 bases of the forward read represent the UMI, followed by a common linker with the sequence “TATA”, followed by the 12bp random priming sequence. It is recommended to transfer the UMI sequence information to the read header and trim the first 22 bases from each read with UMI-TOOLS or custom scripts. The same software can be used to remove PCR-duplicates after the alignments.
Please also the 3’Tag-Seq data analysis recommendations and this note on working with degraded RNA samples.
Comprehensive Batch-Tag-Seq info for download with UC pricing.
Primer and adapter-dimer contamination in sequencing libraries can lead to serious problems like barcode switching (also called barcode hopping). Thus, these short molecules should be removed from the libraries as soon as traces of them become visible on the Bioanalyzer or equivalent. Please note that the Bioanalyzer uses double-strand-specific fluorescent dyes which have a very low affinity to single-stranded primers. Thus, the Bioanalyzer assay severely underestimates the true concentration of free primer molecules.
- Low concentrations of free primers and adapter-dimers can be removed with a bead cleanup e.g. adding 1x the original volume in Ampure XP beads (or equivalent).
- The more stringent option for primer removal is an Exonuclease VII single-strand digest (e.g. with this ExoVII) at 37C for 20 minutes using 1 ul enzyme and the accompanying buffer; followed by a bead cleanup with 1.6 x the original volume in Ampure XP beads. This will not remove primer-dimers.
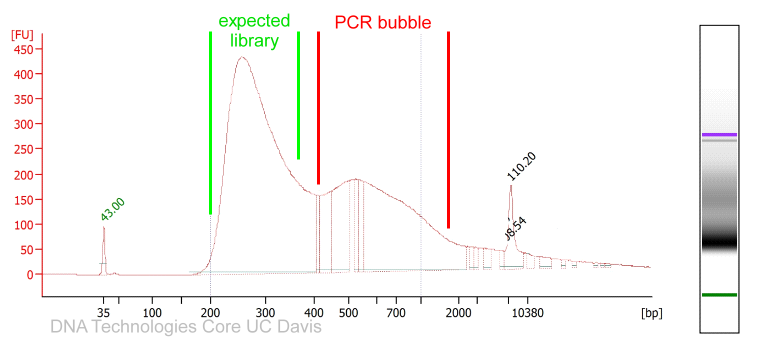
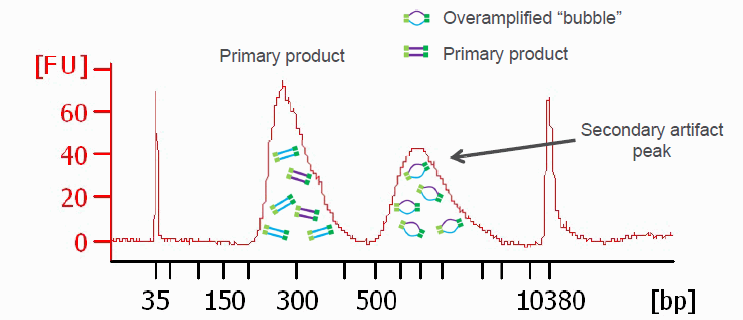
PCR amplified sequencing libraries frequently display library molecules seemingly about twice the excepted size or even bigger. In most cases, this phenomenon is caused by over-amplification of the libraries. These PCR artifacts do occur in cases the PCR reactions run out of essential reagents – in most cases the PCR primers will be exhausted. If primers are no longer available the PCR products will anneal to each other (the sequencing adapter sequence will be the by far most common sequences available). The resulting annealing products are often called “PCR-bubbles” and are partly double-stranded and partly single-stranded; thus they migrate considerably slower on agarose gels as well as on Bioanalyzer assays. Please see below.
Since these artifacts are merely annealing products, the resulting libraries are perfectly sequence-able. However, the quantification of such libraries by fluorometry will not be precise since the dyes used for these measurements are specific for double-stranded DNA molecules and PCR bubbles contain considerable amounts of single-stranded DNA that will not be measured. The PCR bubbles can be removed by amplifying the library one more time with a single cycle of PCR (a so-called “Reconditioning PCR“). For this PCR you could use standard Illumina P5 and P7 primers (please see below). The complementary sequences should be located at the very ends of all Illumina sequencing library molecules. In most cases PCR bubble artifacts can not be removed by SPRI bead size selections or Blue Pippin size selections; if necessary, a “Reconditioning PCR” is the best option.
However, to avoid unnecessary complexity loss of the library and introduction of polymerase errors, it would be best to optimize the library preparation protocol for a lower number of PCR cycles beforehand.
Reconditioning PCR protocol:
The reconditioning PCR uses standard Illumina P5 (5′-AATGATACGGCGACCACCGAGATCT-3′) and P7 (5′-CAAGCAGAAGACGGCATACGAGAT-3′) PCR primers which can be ordered as desalted DNA oligos.
Create a 10x concentrated primer mix at 20 μM each of these primers in EB buffer.
Add 1 to 4 μl previous PCR product, 2 μl 10x primer mix, 3 to 7 μl water for a total volume of 10 μl, then add 10 μl Kapa HiFi 2x Hotstart PCR master mix and pipette up and down several times.
Use the following cycling parameters: Initial denaturation 98°C 45 sec, denaturation 98°C 15 sec, annealing 60°C 30 sec, extension 72°C 30 sec, final extension 1 min.
Perform a standard Ampure XP/SPRI bead cleanup (e.g. with beads at 1.2x the sample volume) before analyzing the product by microcapillary electrophoresis.
Another graphic illustrating PCR bubbles (source Illumina Inc.). Please also see: https://support.illumina.com/bulletins/2019/10/bubble-products-in-sequencing-libraries–causes–identification-.html
Illumina sequencers using the patterned flowcell technology (NovaSeq X, iSeq, NextSeq 2000, HiSeq 4000 &X Ten) can show an increased rate of barcode switching events. These artifacts are enabled by the exclusion amplification chemistry used on these sequencers; IIlumina calls the artifacts “index hopping”. Please see the index hopping information from Illumina here and in this video from Illumina. We have also have posted additional information here.
Please note that the AVITI sequencers, the MiSeq sequencer and NextSeq 500 chemistries are not sensitive to index hopping. For these sequencers single-indices and combinatorial indexing are fine.
To best avoid barcode switching read mis-assignments on new Illumina sequencers two measures are recommended:
- The use of uniquely dual-indexed (UDI) adapters. Illumina has collected information on the design of such adapters here. This adapter design employs unique barcodes for both index 1 and index 2 for each of the pooled libraries. Thus, 96 samples will use 96 different index 1 adapter sequences and 96 different index 2 adapter sequences.
- The efficient removal of any free primers and adapter-dimers from the libraries
The use of UDI adapters is highly recommended especially for the NovaSeq. Hereby, matching i5 and i7 indices per library must be avoided. UDI adapters can also be used on the MiSeq and the NextSeq, but they do not offer any significant advantages on these sequencers that employ the bridge-amplification chemistry. Further, any traces of free primers, primer-dimers, and adapter dimers should be removed from the sequencing libraries or the pools.
Commercial sources of UDI adapters: TruSeq-style uniquely indexed adapters are available from both Illumina and BiooScientific. Qiagen, NEB, and NuGEN are also supporting their library prep kits with optional UDI adapters. Nextera style indices need to be custom ordered from oligo vendors.
The DNA Technologies Core has 96-plex UDI adapter sets in stock that can be added to sequencing libraries by PCR. These barcode sets are available for both Nextera and TruSeq adapter designs. Please note that for TruSeq style libraries one will need to ligate a shortened and index-less stub-adapter instead of a standard Illumina adapter. The indices are then added after the cleanup of the ligation reaction by PCR.
Sequencing libraries prepared by the DNA Technologies Core:
The DNA Technologies Core uses UDI adapters for all library prep protocols that are compatible with dual indexing (e.g. DNA-Seq, RNA-seq, 3′-Tag-Seg, WGBS-Seq, …). The Core also makes sure to remove any traces of free primers, primer-dimers, and adapter-dimers.
- The fragment lengths should be consistent and best be between 100 and 300 bp (up to 400 bp for the majority of molecules is acceptable). Consistent fragment lengths can best be achieved on a Covaris style closed tube sonicator. We recommend avoiding probe sonicators.
- Please make sure to run the input controls on a Bioanalyzer or agarose gel beforehand, and email us an image of these.
- Sequence one “input control” per cell line/sample type.
- Analyze at least two biological replicates.
- We highly recommend verifying the enrichment of your regions of interest (e.g. promoter regions) vs. the control samples by qPCR, before submitting the samples for sequencing.
- For highest accuracy data we can now generate sequencing libraries with UMI-bearing sequencing adapters. UMIs (Unique Molecular Identifiers) allow the accurate detection and removal of PCR duplicate reads. This approach is especially recommended for low-input samples. The first nine bases of the forward and reverse reads will contain UMI sequences.
The required read number per sample will vary from target to target. For the study of point source transcription factors the ENCODE project recommends analyzing at least 20 million (uniquely mapping) reads (http://genome.cshlp.org/content/22/9/1813.long#boxed-text-2). Depending on the quality of your preps, perhaps 75% of the reads can be expected to be uniquely mapping. ENCODE tends to err on the high side with their recommendations. Thus, about 20 million read pairs per sample should be acceptable, but this is likely the minimum number.
Zhang et al. 2016 have studied the impact of the sequencing run types on ChIP-seq data analysis. Their data indicate that paired-end sequencing data provide significant advantages of single-end sequencing in ChIP-seq.
CUT&RUN sequencing might be a better alternative:
CUT&RUN sequencing (Skene & Henikoff 2017) is a faster protocol that for almost all applications is a more sensitive alternative requiring much lower cell numbers. CUT&RUN is suitable for studying histone modifications, transcription factors, and co-factors. In addition to lower input requirements CUT & RUN experiments also afford reduced read numbers (4 to 8 million read pairs per sample).
References:
Landt et al. 2012: ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Research 22: 1813-1831
Bailey et al. 2013: Practical Guidelines for the Comprehensive Analysis of ChIP-seq Data. PLOS Computational Biology https://doi.org/10.1371/journal.pcbi.1003326
Skene & Henikoff 2017: An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. https://doi.org/10.7554/eLife.21856
Zhang et al. 2016: Systematic evaluation of the impact of ChIP-seq read designs on genome coverage, peak identification, and allele-specific binding detection. BMC Bioinformatics volume 17, Article number: 96
MACS — Model-based Analysis of ChIP-Seq https://taoliu.github.io/MACS/
https://hbctraining.github.io/Intro-to-ChIPseq/lessons/05_peak_calling_macs.html
We have currently 96 indices are available and can pool 96 RNA-seq or genomic sequencing libraries. Bioo Scientific offers NEXTflex barocde sets allowing the pooling of up to 384 libraries. If you are planning to use homebrew versions of indices please consult with us first, as reduced complexity from incorrectly designed indices may cause failures when sequencing your sample.
If you have access to fluorometric DNA quantification and a Bioanalyzer (or equivalent), library pooling is not difficult. We offer the pooling of sequencing libraries for a small fee. For sequencing libraries generated by the Core, pooling is included in the library preparation service.
Prerequisites for the pooling of customer libraries are:
- all libraries were generated using the same protocol and are PCR amplified
- the library fragment sizes have to be similar for all libraries * (and within Illumina specs) as demonstrated by Bioanalyzer traces (or gel images if correct balancing is not that critical)
- have uniquely indexed adapters
- all libraries have DNA concentrations in the same range
- PCR-amplified libraries can be quantified based on fluorometric measurements (e.g. Qubit), but PCR-free libraries are best quantified by qPCR.
Library pooling requires precise pipetting of very small volumes. Even with the best libraries, there will be some imbalances. However, and we can’t work magic with variably sized samples.
* The clustering efficiency of Illumina sequencing libraries varies with the fragment lengths. Shorter molecules are more mobile and will always cluster preferentially compared to longer molecules (smaller molecules will “win the race” to the flowcell surface oligos). Thus, accurate pooling is impossible when combining libraries of varying lengths and we can’t vouch for the results. In some cases, it is advisable to size-select the libraries stringently before quantification and pooling.
For sequencing libraries generated by the Core, pooling is included in the library preparation service.
We suggest the following procedure when pooling libraries yourself:
- verify that Bioanalyzer traces of your libraries show the same fragment size distribution
- quantify each library by fluorometry (Qubit or plate reader)
- if necessary dilute some of the highly concentrated libraries (to bring them in line with the others)
- re-quantify the newly diluted libraries (Qubit)
- Under the precondition, that all libraries show very similar fragment size distributions there are in principle two options:
#1 Dilute all libraries to the same concentration, re-quantify the libraries, then pool the same volumes. This option is more laborious than #2, but allows more precise re-pooling if needed (since all libraries have similar concentrations at least).
#2 Pool the same a amounts of each library based on the first round of quantifications. This means pooling varying volumes. As long as the libraries are consistent in length this could mean pooling the same amounts in ng. If the length is a bit variable one should pool the same number of femtomoles for each library; the femtomole amount is calculated by multiplying the concentration (in nM) by volume (in ul). The molarity calculation will consider the fragment lengths and compensate for varying length. But see tip 3) below !!! - Quantify the resulting pool by Qubit to verify that it has the expected concentration (we will quantify once more by qPCR before sequencing)
Please note that the combined library concentration of the pool should be 5 nM or higher (Illumina sequencers) or 16 nM or higher (AVITI); this means that the concentration of individual libraries in the pool can and will be considerably lower.
You can get trained and use a Qubit in our lab: http://dnatech.genomecenter.ucdavis.edu/qubit-fluorometer/
Three more tips for library pooling:
1) Best pipette volume ranges that allow the pipetting to be reproducible and accurate while still saving some library. For example aiming for a volume range from 3 ul to 10 ul.
Likely you will pool much more library than is needed in the end for sequencing.
2) In case you chose option #2 above and your library concentrations vary more than +- 60%, it is often helpful to work with two sub-pools: One for the higher concentration samples and one pool for the lower concentration samples.
The example numbers here are arbitrary. Under the precondition, that all libraries show very similar fragment size distribution, one would for example pipette 100 ng of each high-concentration library into one tube (Pool A) and 20 ng of each low-concentration library into another tube (Pool B). Vortex mix and spin down each of the tubes twice.
For the final combined pool one could then use the complete volume of Pool B and add one fifth of the total volume of Pool A. Vortex and spin down the combined pool twice. The final combined pool thus should contain 20 ng for each of the libraries aiming for balanced sequencing data. (Please note that the library amounts and pooling ratios mentioned here are only examples and that you have to choose appropriate ratios based on the library concentrations of your experiment).
3) If the fragment length distribution of the libraries is variable, one should try to adjust for these length differences (convert the library concentrations into molarities for this purpose). Further consider that shorter library molecules will be faster and cluster preferentially. To generate similar read numbers, one has to pool more library molecules for longer library molecules. Obviously more guessing and a bit of gambling will be involved in this case and pooling will be less accurate.
Strand-Specific RNA-Seq Libraries
RNA-Seq (conventional) after Poly-A enrichment or ribodepletion:
By default we generate strand-specific RNA-seq libraries. Strand-specific (also known as stranded or directional) RNA-seq libraries substantially enhance the value of an RNA-seq experiment. They add information on the originating strand and thus can precisely delineate the boundaries of transcripts in regions with genes on opposite strands.
There are several ways to accomplish strand-specificity. We incorporate dUTP during the second-strand synthesis of the cDNA. The dUTP containing strand will not be amplified by the proofreading polymerase used for library amplification, thus preserving the strand information for RNA-seq.
For single-end sequencing, the resulting data will represent the “anti-sense strand”. When using paired-end sequencing, the forward read of the resulting sequencing data represents the “anti-sense strand” and the reverse read the “sense strand” of the genes (for Trinity transcriptome assemblies the “–RF” orientation flag should be used). Illumina paired-end reads are always inward oriented (with the exception of “jumping” or “mate-pair” libraries).
Tag-Seq:
Tag-seq data are strand-specific and have a “sense-strand” orientation.
small-RNA-Seq/miRNA-seq:
Small RNA-seq data are strand-specific. The forward read of the sequencing data (read 1) is oriented as the reverse complement of the original RNA molecule. For libraries generated with the Revvity (PerkinElmer) Nextflex small RNA kits, the RNA sequence will be both preceded and followed by four bases with a random sequence.
Illumina sequencing libraries are usually generated with Y-adapters. These are partly single-stranded and partly double stranded.
A PCR-free library will thus still contain partly single-stranded regions. These single-stranded regions can lead to several types of Bioanalyzer artifacts. Most commonly the libraries will appear about 70 to 100 nucleotides longer than expected. However, we have also encountered PCR-free libraries that ran as shorter molecules as well as dramatically longer molecules. We have (very rarely) encountered another significant problem: considerable amounts adapter-dimers were not visible on the Bioanalyzer traces of PCR-free libraries.
To accurately QC PCR-free Illumina libraries we recommend the following approach:
– Take a 1 ul aliquot of your library and run a short PCR (e.g. 6 cycles) with this aliquot.
– Clean up the PCR reaction with a spin column ( e.g. Qiagen Qiaquick, Zymo DNA -clean, …); do NOT use Ampure beads.
– Run the cleaned up PCR product on the Bioanalyzer again as well as the original PCR-free library.
The Bioanalyzer trace of the PCR product will represent the true molecule sizes and the true adapter-dimer content the closest.
Is PCR-free library preparation still advantageous?
If we generate PCR-free libraries in our lab, the described additional QC steps for PCR-free libraries will necessitate significant additional costs for the library preparation. Please see the prices for the PCR-free Add-on.
A great alternative to preparing the libraries completely PCR-free is the use of a single PCR cycle instead. This combines the advantages of both: fully double-stranded library molecules can be used for the library QC and there will be no or only an extremely low PCR-bias introduced. Our recommendation is to submit the same amount of DNA sample as for PCR-free library preps (1 ug or more) and then only apply the single cycle of amplification.
In general, the original grave concerns about library PCR amplification (presented in papers from 2011) are no longer very relevant. This is due to the use of modern polymerases that are designed for complex samples like Kapa HiFi, NEB Q5, or QIAseq HiFi polymerase. The previous “standard”, the high-fidelity Phusion enzyme had tremendous disadvantages for complex samples (Quail et al. 2012 Optimal enzymes for amplifying sequencing libraries. Nature Methods volume 9, pages10–11(2012) https://www.nature.com/articles/nmeth.1814 ).