
03 Sample Preparation & Sample Requirements
DNA Sample Integrity:
For Illumina short-read sequencing:
DNA sample integrity should best be QC-ed by agarose gel-electrophoresis and ethidium bromide staining. “Safe” gel-stains such as Gel-Red work just as well.
These stains will make both DNA and RNA visible. RNA will run as an halo-like smear in the range 50 to 300 bp.
We suggest a 1% agarose gel and a ladder marker that best includes a 20 kb band like the GeneRuler 1kb Plus DNA ladder from Thermo Scientific. Please load about 40 to 100 ng DNA for each sample. Other conditions can work as well.
The agarose gel image will show the presence or absence of RNA contamination and provide the best information on potential sample degradation.
Please email us an agarose-gel image before shipping the samples in case of any concerns. Please always ship a copy of the agarose-gel image together with the samples.
For PacBio or Nanopore long-read sequencing:
HMW-DNA samples should be QC-ed via pulsed-field gel electrophoresis (PFGE) or field-inversion gel electrophoresis (FIGE). We can carry out this QC for you. The Femto-Pulse will instrument enables capillary FIGE with ultra-low input amounts and will provide a digitized data analysis (similar to the Bioanalyzer for short molecules).
If you do not have access to these technologies, we suggest running a longer conventional agarose gel (as described above) to get a first idea about the sample quality before shipping the samples to us for a FIGE analysis.
Please always ship a copy of agarose-gel images together with the samples.
DNA Sample Purity:
DNA sample purity has to be determined via spectrometry. Please see the sample requirements page for the recommended values for your protocol. It is certainly helpful to also record the entire UV absorption spectrum as it provides additional information. For DNA samples the 260/230nm ratio should be >2 and the 260/280nm ratio 1.8-2.0 .
Please also see:
Which DNA isolation protocols do you recommend for Illumina sequencing?
How should I purify my samples? How should I remove DNA or RNA contamination?
Do you offer DNA isolations and RNA isolations as a service?
How do I prepare DNA samples for RR-Seq (reduced representation sequencing)?
- Spin column DNA isolation kits are available from multiple vendors including Qiagen, Zymo, Omega Biotek, Sigma, and Norgen Biotek e.g. Qiagen DNeasy Blood & Tissue kit with added RNAse A (RNase A 100 mg/ml; cat. no. 19101).
- The Qiagen DNeasy Blood & Tissue kit (with added RNAse A) is also the default kit for bacterial isolate DNA extractions. The kit comes with dedicated bacterial protocols.
- Some vendors also offer DNA isolation kits in a 96-well spin-plate format for large sample numbers (e.g. Qiagen, Zymo).
- Only use a protocol that includes an RNase digestion step to remove any contaminating RNA; RNA can inhibit the DNA sequencing library preparation.
- Plant samples will require a dedicated kit that includes a lysis buffer designed to capture harmful plant chemicals like phenols (e.g. Qiagen DNeasy Plant). Without protective additives in the lysis buffers, plant chemicals will damage the DNA.
- Similarly, soil samples are rich in inhibitors of enzymatic reactions. Dedicated protocols and kits that can remove such chemicals (e.g. DNeasy Powersoil Pro) and are highly recommended.
- If accurate quantification of the resulting DNA samples is required, absolutely avoid any protocols that employ the chemical CTAB. Spin column protocols are usually CTAB-free.
- To achieve the cleanest DNA isolation, only use at most half the sample amount of the maximum recommended by the manufacturer.
- Spin-column isolation tips: perform the “optional” steps described in the manufacturers manual. Always perform at least two spin column washes (with the kit wash buffer) after binding of the sample to the column matrix. Add a short “dry spin” of the column after the washes and before the elution buffer addition to avoid any carryover of the ethanol wash buffer. Extend the incubation times for elution of DNA samples from spin columns to at least 5 minutes – or perform two consecutive elutions instead.
- NEVER use heparin as an anticoagulant for blood samples destined for DNA or RNA sequencing. EDTA (preferred) or citrate anticoagulants should be used. Heparin co-purifies with nucleic acids and inhibits multiple types of enzymes like polymerases and ligases.
DNA Sample QC:
- After extraction the DNA sample purity has to be determined via spectrometry (e.g. Nanodrop). Please see the sample requirements page for the recommended values for your protocol. It is certainly helpful to also record the entire UV absorption spectrum as it provides additional information. For DNA samples the 260/230 nm ratio should be >2 and the 260/280 nm ratio 1.8-2.0.
- To assess the DNA sample integrity and verify the removal of RNA, DNA samples should best be analyzed by agarose gel electrophoresis, see: How should I QC my genomic DNA samples before sequencing? Please email us an agarose gel electrophoresis image with the DNA samples. For spin-column protocols the DNA fragments should be longer than 10 kb or 15 kb. Shorter fragments indicate DNA damage before the DNA isolation; please inquire with us in such cases.
Please also see:
How should I QC my genomic DNA samples before sequencing?
How should I purify my samples? How should I remove DNA or RNA contamination?
Do you offer DNA isolations and RNA isolations as a service?
How do I prepare DNA samples for RR-Seq (reduced representation sequencing)?
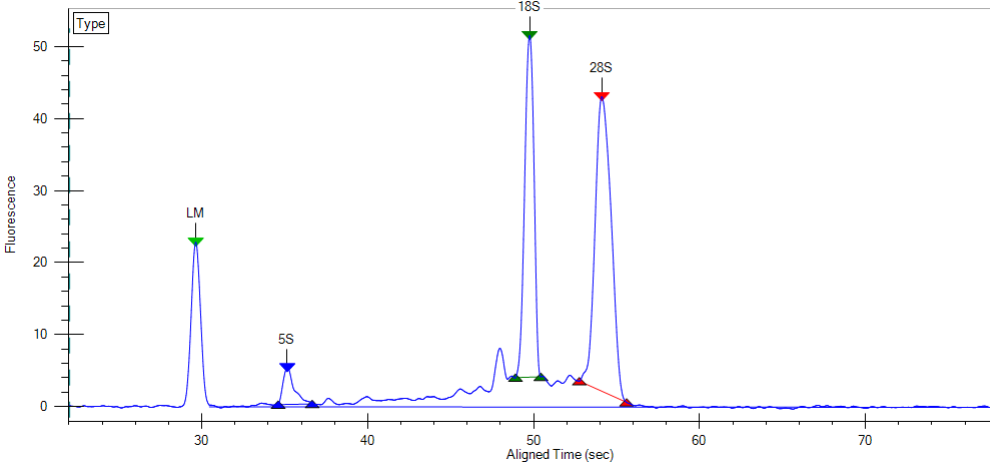
Assessing the integrity of RNA samples with the Bioanalyzer can be time-consuming and expensive since each run takes an hour and only 12 RNA can samples can be run.
To make RNA QC more convenient and affordable we will be running one or more batches of RNA samples weekly on the high-throughput LabChip GX.
For UC Davis labs the cost per sample will be $5 (with a minimum of $10).
In order to generate usable data:
- Provide sample names with an implicit order. The plots of the traces will identify the well position not sample names.
- We will not adjust sample concentration or volumes. It is your responsibility to meet the sample requirements.
- Submit total RNA samples in a well-sealed 96-well plate or in strip tubes (please see this page for examples) and a filled out QC submission form.
- For large sample numbers make sure that the plates are clearly labeled. Please use the QC submission form simply as a cover sheet in this case, listing the plate names and the number of samples for each. Samples should be filled into plates in column order from A1 to H1, then A2 to H2, etc. …
- Each RNA sample needs to have a volume 2 ul to 6 ul and contain 30 ng to 250 ng total RNA (this is the amount, not the concentration).
- Glycogen can interfere with the RNA QC and should be avoided (it can interfere with both spectrometry as well as capillary electrophoresis on the LabChip and the Bioanalyzer).
The LabChip GX will generate traces like the one below and RNA Quality Scores which are similar to the RIN scores provided by the Agilent Bioanalyzer and can be used interchangeably.
Please note that the capillary electrophoresis will be of lower resolution compared to the Bioanalyzer, but “good enough”. There is also no dedicated quality score algorithm for plant samples on the LabChip. The scores it produces are still realistic.
Unfortunately, we cannot demultiplex inline barcodes.
Depending on sequencer model, the second index (called i5 index) will be read in different orientations. The correct adapter sequence information for the sample sheets is usually provided for both options by kit manufacturers. The two sequences are reverse complements of each other.
- The Forward Strand Workflow (previously known as Workflow A) sequences are required for MiSeq and AVITI.
- The Reverse Strand Workflow (previously Workflow B) sequences are required for NovaSeqs and NextSeq.
BTW, the AVITI demultiplexing software is smarter and can figure out the correct orientation on its own. Either orientation will be fine for AVITI submissions.
Here is the complete information on this topic from Illumina: https://support-docs.illumina.com/SHARE/IndexedSeq/indexed-sequencing.pdf
To check if the genome of your species of interest is suitable for Optical Genome Mapping on the Bioanao Saphyr, you should check the distribution of labeling sequence motifs. For this purpose, Bionano provides in silico digestion tools with the “Label Density Calculator” program. “Bionano Access” also has such a feature. Both programs are available from this webpage: https://bionanogenomics.com/support/software-downloads/
Quality and quantity of DNA and RNA is critical for high quality sequencing output. Please make sure your DNA is not degraded and is free of RNA contamination. RNA samples should always be assessed on the bioanalyzer for the absence of gDNA contamination (can be removed with DNaseI treatment followed by a column clean-up; e.g. Zymo “RNA Clean and Concentrator”) and degradation. Preferentially determine the concentrations of your DNA and RNA samples using fluorometry (e.g. with a Qubit or plate reader). The sample purity should be assessed by spectrophotometry (e.g. Nanodrop). Please see this page for a comprehensive table of sample requirements for sample QC, library preps, or your self-made libraries. Please see the Library Prep Page for details on the library prep processes. For submission information, including submission forms and shipping details, please visit the Sample Submission & Scheduling page. If you are submitting DNA for PacBio libraries, please follow the PacBio Guidelines for Shipping and Handling.
The Real-time PCR core can carry out DNA as well as RNA extractions for you.
What type of samples are recommended for the isolation of HMW-DNA? (for Long-Read Sequencing)
Please see the information in this PDF that we wrote originally for the California Conservation Genomics Program (CCGP).
It contains recommendations for the collection of samples for both DNA and RNA isolations for the purpose of reference-quality genome assemblies and gene annotations.
Sample Collection Recommendations for Long-Read Sequencing and Gene Annotations
What type of samples are recommended for RNA isolations for gene annotations?
Please see the information on page three of this PDF that we wrote originally for the California Conservation Genomics Program (CCGP).
It contains recommendations for the collection of samples for both DNA and RNA isolations for the purpose of reference-quality genome assemblies and gene annotations.
Sample Collection Recommendations for Long-Read Sequencing and Gene Annotations
The sample amount requirements are chosen to ensure both, high-quality data and efficient processing.
Most of the library prep protocols will generate sequenceable libraries with lower input amounts than request, often requiring additional PCR cycles.
Processing often low-input samples often requires additional handling and QC steps. Thus, additional custom processing costs may apply.
Please contact us before submitting such samples. When working with sample amounts lower than recommended, you will generally run the risk of introducing biases and noise into your data. This may or may not be an acceptable trade-off for your specific project.
The sample integrity requirements are chosen to ensure the generation of high-quality data. Please contact us before submitting such samples. It may be possible to use an alternative protocol that tolerates some sample degradation.
When working with samples of lower integrity than recommended, you will generally run the risk of introducing biases and noise into your data. This may or may not be an acceptable trade-off for your specific project.
We can certainly work with degraded samples if requested, but we cannot vouch for the quality of the resulting data.
Processing of low-integrity samples often requires additional handling and QC steps. Thus, additional custom processing costs may apply.
Bead based sample cleanups (e.g., Ampure XP, RNAClean XP) and spin column-based protocols (e.g., Qiagen, Zymo, NorgenBiotek) tend to be the most efficient ways to remove chemical contaminants. For genomic DNA samples to be sequenced on Illumina sequencers, we suggest spin columns since DNA treated this way will always dissolve well and completely.
Please test for chemical contamination by spectrophotometry (e.g., Nanodrop), concentrations should be measured by fluorometry instead (Qubit, Quantus, plate reader, …) :
- Please see this guide from the University of Arizona on the interpretation of Nanodrop data. Skewed absorption ratios indicate that there is chemical contamination, but not precisely which contaminant and if it will be deleterious or not,
- The 260/230 nm and 260/280 nm absorption ratio measurements are most frequently used to assess purity. Please see the sample requirements page for the recommended values for your protocol. However, it is certainly helpful to also record the entire UV absorption spectrum as it provides additional information. For RNA the 260/230nm ratio should be >1.5 and the 260/280nm ratio 1.8-2.1; For DNA the 260/230nm ratio should be >2 and the 260/280nm ratio 1.8-2.0 .
- In case the absorption ratios are skewed, it is often worth checking if any alcohol was carried over from the spin column or bead washes. Any organic substance, including ethanol, will skew the 260/230 nm ratios. One can vent the open sample tube (for example for 20 minutes) on the lab bench and measure again afterwards to see if the contamination has disappeared.
- The spectrophotometer ratios themselves become easily misleading at very low DNA or RNA concentrations (10 ng/ul or less). In these cases the nucleic acid samples contribute very little to the signal and the slightest contamination dominates the readings. Please record the absorption spectra.
Multiple protocols are available to remove DNA or RNA contaminants. Please find our suggestions for affordable solutions for Illumina sequencing below.
RNA samples need to be DNA-free. The RNA isolation protocol should always include a DNase digestion step; in problematic cases use RNA-clean & concentrator kits with DNase. On an agarose gel, DNA contamination will be visible as a smear or band of fragments considerably larger than the RNA (>10 kb). On Bioanalyzer RNA-chips, DNA contamination will be visible in the size range 4 kb to 10 kb.
If you are using a Trizol protocol for the RNA extractions we would highly recommend cleaning the samples afterwards with a spin column kit (e.g. RNA-clean & concentrator kits) to remove any phenol traces.
Please note that the additional column cleanup is mandatory for RNA samples isolated from blood PAXgene or Tempus tubes (for blood sample preservation) or with the accompanying PAXgene and Tempus RNA isolation kits.
DNA samples need to be RNA-free. The DNA isolation protocol should always include an RNase digestion step; in problematic cases we recommend using RNase I (e.g. add 1 ul RNAse I to your sample and incubate at 30 degrees C for 20 minutes). RNase I does not require a special buffer (it works in TE buffer). For the removal of the RNase I, Ampure XP beads (or similar) or DNA-clean & concentrator kits will work fine (we suggest extending incubation times for elutions from the columns to at least 5 minutes or to perform two elutions). Do NOT try to inactivate the RNAse by heating (the NEB manual suggests heating to 70°C – this will already denature DNA dissolved in water or EB buffer and introduce biases in the library preparation!
DNA samples can be QC-ed easily by agarose gel electrophoresis and ethidium bromide staining. The stain will make both DNA and RNA visible. RNA will run as an halo-like smear in the range 50 to 200 bp.
For the removal of chemical contaminants solid-phase paramagnetic bead cleanups (SPRI-beads) are a solution suitable for high-throughput processing. The first such products were Ampure XP (for DNA) and RNAClean XP from Agencourt/Beckman. Many companies are now selling lower-cost versions, for example MagBioGenomics DNA beads and RNA beads.
We can recommend this EdgeBio magnetic plate for bead cleanups in 96-well plates.
- For spin-column cleanups: Please perform the optional steps described in the manual. Always perform at least two spin column washes (with the kit wash buffer) after binding of the sample to the column matrix. Also, add a short “dry spin” of the column after the washes and before the elution buffer addition to avoid any carryover of the ethanol wash buffer.
- We suggest extending incubation times for elutions of DNA samples from spin columns to at least 5 minutes – or to perform two consecutive elutions instead.
- NEVER use heparin as an anticoagulant for blood samples destined for DNA or RNA sequencing. EDTA (preferred) or citrate anticoagulants should be used. Heparin co-purifies with nucleic acids and inhibits multiple types of enzymes like polymerases and ligases.
- Avoid using glycogen as co-precipitant.
DNA samples for long-read sequencing library preparations or also 10X genomics linked-reads have to be exceptionally pure. Please see the sample requirements. For difficult DNA samples, especially all plant DNA samples with hard-to-remove contaminants (e.g. some polysaccharides), we recommend to carry out a high-salt/phenol/chloroform cleanup (please see this protocol) . Please note that this protocol often leads to a loss of 50% of the sample. Alternatively the the BorealGenomics Aurora instrument (discontinued but still available, please inquire) can be used. This process is slower though (only a single sample can be processed per day or two days) and is accompanied by similar or greater samples loss.
At the moment we only carry out high-molecular-weight DNA (HMW-DNA) isolations for the purpose of 10X Genomics and Nanopore sequencing. Please inquire with Ruta Sahasrabudhe, PhD.
We do not offer DNA isolations for Illumina sequencing and RNA isolations at the moment. However the Taqman Core does. Please contact the Real-time PCR and Research Diagnostics Core (also known as the Taqman Core). They carry out nucleic acid isolations from a wide variety of tissues for both plant, animal, bacterial , and fungal samples. The Taqman Core manager Samantha Barnum and her team have many years of experience in the extraction of sequencing-worthy DNA and total RNA samples. Please note that for sequencing purposes the ‘Qiagen Nucleic Acid Extraction’ option should be selected – this protocol generates the highest quality material for Illumina sequencing for both DNA and RNA. The same protocol may also be suitable for PacBio sequencing, but is only recommended for bacterial samples.
We encourage scheduling your DNA and RNA extraction services directly with the Taqman Core and to mention that the samples are designated for sequencing. Please contact the Taqman Core manager Samatha Barnum for technical details/sample requirements.
There are multiple valid protocols available for amplicon sequencing on Illumina systems. Here we describe one of many options: A two-step PCR protocol to generate complete sequencing libraries.
This protocol has the advantage that it does not require custom sequencing primers and that the barcode-indexing oligos can be re-used for multiple different amplicons and future projects. We suggest to follow a “16S amplicon” protocol that was explicitly designed by Illumina to be adaptable to other targets (please see the full protocol and pages 3 and 4 here).
Once you have designed the oligos as described in the Illumina protocol (forward overhang plus your sequence-specific primer as well as reverse overhang plus sequence-specific primer), we suggest checking these sequences on the IDT oligoanalyzer ( https://www.idtdna.com/calc/analyzer ) for secondary structures. It is advisable to avoid any sequences that generate a Delta G smaller than -9 for any of the structures.
There is no need to purchase an Illumina Nextera index kit. The sequences for the index primers (26 i7 index 1 sequences; 18 i5 index 2 sequences) are available on pages 7 and 8 here. These indices allow for the combinatorial sequencing of up to 468 samples. When ordering oligos please use the index sequences in the “Bases in Adapter” columns. The oligos are used for standard PCR reactions. Thus, low-cost desalted oligos can be ordered for this purpose anywhere and will work just fine. We strongly recommend using plates with single-reaction aliquots of these index primers for your experiments to make sure that index primer stocks cannot become contaminated.
Your first round PCR amplicon products will have universal tails/tags/overhangs on both ends. Since you can use dual indexes, you could order for example 5 index oligos with i5 indexes and 5 index oligos with i7 indexes and have 25 usable barcode combinations for your project. If you are using single indices they have to be i7 (P7 adapter) indices. However, for HiSeq 4000 and NovaSeq sequencing you should use uniquely-dual-indexed (UDI) barcode combinations.
The first round PCR primer designs use Nextera-style tag sequences (overhang sequences) and look like this:
Forward overhang P5-tag: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-[locus-specific sequence]
Reverse overhang P7-tag: 5’ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-[locus-specific sequence]
The second round PCR primers are Nextera-style index primers – i5 and i7 indicate the location of the barcode index sequences:
P5-PCR index primer: 5’ AATGATACGGCGACCACCGAGATCTACAC[i5]TCGTCGGCAGCGTC
P7-PCR index primer: 5’ CAAGCAGAAGACGGCATACGAGAT[i7]GTCTCGTGGGCTCGG
Please optimize the conditions of the first round PCR to avoid primer-dimer generation. The PCR reactions should be cleaned up with Ampure XP beads (or similar) and resuspended in EB buffer.
Once you have verified (via agarose gel electrophoresis) that the PCR products for all samples are clean and of about the same and expected size, the samples should be pooled equimolarly. We suggest to quantify the samples via fluorometry (Qubit or plate reader) for accurate pooling.
In case you are targeting only a single amplicon, it helps to create sequence diversity by adding a set of PCR primers with added diversity spacer “N” bases (or defined bases; up to seven of them) between the overhangs for both forward and reverse primers (Fadrosh et al. 2014, Wu et al. 2015). The resulting set of primers should be pooled in equimolar ratios and used for the first round of PCR.
The original Illumina design looks like this: overhang+locus-spec. sequence (no spacer):
5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐[locus‐specific sequence]
Complementary stagged spacer versions of this oligo would be:
One spacer base added: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐X-[locus‐specific sequence]
Two spacer bases added: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐XX-[locus‐specific sequence]
Three spacer bases added: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐XXX-[locus‐specific sequence]
Four spacer bases added: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐XXXX-[locus‐specific sequence]
Five spacer bases added: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐XXXXX-[locus‐specific sequence]
Six spacer bases added: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐XXXXXX-[locus‐specific sequence]
Seven spacer bases added: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐XXXXXXX-[locus‐specific sequence]
Knowing the locus-specific sequence one can certainly be smarter and make the two “N”s directly before the locus‐specific sequence different from the first two bases of the locus‐specific sequence (Fadrosh et al. 2014). If pooling amplicons for multiple targets (more than 8) there is no advantage using diversity spacers.
Some downstream programs might require the removal of the diversity spacers. dbcAmplicons can demultiplex the data as well as trim/remove the diversity spacer.
Fungal ITS: Illumina has published a second version of this protocol, modified to sequence and study fungal ITS sequences.
Qiagen offers a commercial amplicon prep kit for multiple 16S regions and ITS for which they have perfected the diversity spacer approach described above. This kit eliminates the need for PhiX spike-ins.
A much more detailed protocol for 16S and other amplicon sequencing is available here: Gohl et al. 2016
Please see this page for the library requirements for sequencing (http://dnatech.genomecenter.ucdavis.edu/sample-requirements/). The above protocol will generate a surplus of library material.
RNA samples should best be shipped on dry ice. Please only ship with courier services (FedEx, UPS, DHL).
For longer transports (e.g. from South America) we also had very good success with RNA samples shipped dry at room temperature (after LiCL/ethanol precipitation and ethanol washes; see protocol below).
Please mark the position of the pellet on the tubes in this case!
The protocol below is modified from here: http://www.paralog.com/wiki/?EthanolPrecipitation
- Add 1/10th volume of 8M LiCl to your RNA sample and mix.
- Add 3 volumes of room temperature 100% ethanol (based on the aqueous volume before the addition of LiCl) and mix thoroughly.
- Incubate at -20C for 20 minutes or ON.
- Vortex quickly. Mark the side of the tube that will be positioned towards the outside of the centrifuge rotor (the side the pellet will be located).
- Centrifuge at a minimum of 12000g for 15 minutes in a cooled centrifuge (4C).
- Decant the supernatant.
- Add 500 μl room temperature 75% ethanol and rinse the tube by gentle inversion.
- Respin at 12000g for 2 minutes.
- Decant the 75% ethanol.
- Spin down briefly and pipette aspirate the remaining 75% ethanol.
- Repeat the 75% ethanol wash (the previous five steps)
- Allow the pellet to air dry. The sample is now ready for shipping at room temperature.
The samples can then be resuspended in an appropriate volume of molecular biology grade water (best for short term storage and library preps), EB buffer, or TE buffer (only long term storage). Please let us know how much RNA sample to expect per tube.
Please see this FAQ for RNA isolation tips: http://dnatech.genomecenter.ucdavis.edu/faqs/which-protocols-or-kits-do-you-recommend-for-rna-isolations-from-human-and-animal-samples/
- The fragment lengths should be consistent and best be between 100 and 300 bp (up to 400 bp for the majority of molecules is acceptable). Consistent fragment lengths can best be achieved on a Covaris style closed tube sonicator. We recommend avoiding probe sonicators.
- Please make sure to run the input controls on a Bioanalyzer or agarose gel beforehand, and email us an image of these.
- Sequence one “input control” per cell line/sample type.
- Analyze at least two biological replicates.
- We highly recommend verifying the enrichment of your regions of interest (e.g. promoter regions) vs. the control samples by qPCR, before submitting the samples for sequencing.
- For highest accuracy data we can now generate sequencing libraries with UMI-bearing sequencing adapters. UMIs (Unique Molecular Identifiers) allow the accurate detection and removal of PCR duplicate reads. This approach is especially recommended for low-input samples. The first nine bases of the forward and reverse reads will contain UMI sequences.
The required read number per sample will vary from target to target. For the study of point source transcription factors the ENCODE project recommends analyzing at least 20 million (uniquely mapping) reads (http://genome.cshlp.org/content/22/9/1813.long#boxed-text-2). Depending on the quality of your preps, perhaps 75% of the reads can be expected to be uniquely mapping. ENCODE tends to err on the high side with their recommendations. Thus, about 20 million read pairs per sample should be acceptable, but this is likely the minimum number.
Zhang et al. 2016 have studied the impact of the sequencing run types on ChIP-seq data analysis. Their data indicate that paired-end sequencing data provide significant advantages of single-end sequencing in ChIP-seq.
CUT&RUN sequencing might be a better alternative:
CUT&RUN sequencing (Skene & Henikoff 2017) is a faster protocol that for almost all applications is a more sensitive alternative requiring much lower cell numbers. CUT&RUN is suitable for studying histone modifications, transcription factors, and co-factors. In addition to lower input requirements CUT & RUN experiments also afford reduced read numbers (4 to 8 million read pairs per sample).
References:
Landt et al. 2012: ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Research 22: 1813-1831
Bailey et al. 2013: Practical Guidelines for the Comprehensive Analysis of ChIP-seq Data. PLOS Computational Biology https://doi.org/10.1371/journal.pcbi.1003326
Skene & Henikoff 2017: An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. https://doi.org/10.7554/eLife.21856
Zhang et al. 2016: Systematic evaluation of the impact of ChIP-seq read designs on genome coverage, peak identification, and allele-specific binding detection. BMC Bioinformatics volume 17, Article number: 96
MACS — Model-based Analysis of ChIP-Seq https://taoliu.github.io/MACS/
https://hbctraining.github.io/Intro-to-ChIPseq/lessons/05_peak_calling_macs.html
The isolation of high-quality DNA and RNA samples from plants can be challenging due to the presence of inhibiting and damaging phytochemicals. Thus, it is not possible to recommend a single protocol that works for all samples. In any case the RNA samples should be DNAse treated, and QC-ed on a Bioanalyzer for sample integrity and via Nanodrop for purity. Please see sample requirements.
For many species and many types of samples the Qiagen RNeasy Plant MiniKits (cat. no. 74903) have been applied successfully. For RNA-seq and Tag-Seq projects this kit has to be used in conjunction with the Qiagen RNase-Free DNase Set (cat. no. 79254) . We recommend to isolate the RNA, then perform the DNAse digestion on the isolated RNA, and then clean up this reaction once more with the RNeasy kit. Alternatively, you could use the Zymo RNA clean & concentrator 5 kit with DNAse (cat. no. R1013) for the DNAse digestion and clean up.
The NEB Monarch Total RNA Miniprep Kit comes with an “RNA Protection Reagent” that can be added during the mechanical disruption of plant samples.
- Always perform at least two spin column washes (with the kit wash buffer) after binding of the lysed sample to the column matrix. Perform the “optional” steps described in the kit manual. Also, add a short “dry spin” of the column after the washes and before the elution buffer addition to avoid carryover of the ethanol wash buffer.
- Avoid glycogen (often used as a co-precipitant).
- To achieve the cleanest RNA isolations only use at most half the sample amount of the maximum recommended by the manufacturer.
Avoiding Batch-Effects:
Both sample storage conditions and details of the RNA-isolation protocols are well-known to introduce technical variations into RNA-seq data. Because of this, it is recommended to:
- Isolate the RNA-samples in one batch.
- If RNA-isolations need to be carried out in several batches, they should be carried out by the same person using the same batch of reagents
- If RNA-isolations need to be carried out in several batches, the samples should be randomized between the RNA isolation batches (worth discussing with a statistician or the Bioinformatics Core).
- The first PCR is carried out with sequence-specific oligos that are tagged on the 5′-ends with universal tags. The sequences are provided below.
Please note that these first-round primers are amino-modified at the 5′-end to prevent the conversion of unbarcoded amplicons into library molecules. Ordering desalted oligos should be sufficient, as long as the sequence-specific part of the oligo is short (e.g. 20 bp). - The second PCR then uses the universal tags to add sample-specific 16 bp barcode indices to both ends of each amplicon.
REVERSE Tag (U2) 5’-/5AmMC6/TGGATCACTTGTGCAAGCATCACATCGTAG-3’
Sample preparation:
If it has been established that a restriction enzyme (e.g. ApeKI) and method are suitable for the species you are working on (please see below), we require the samples for RR-Seq to be submitted in a 96-well plate. One or two wells should remain empty for negative controls. The concentration of the samples should be normalized to 50 ng/ul as assayed by an intercalating dye (fluorometry using a Qubit; Quantus, or plate reader). To ascertain the chemical purity of the samples, the UV absorption ratios should be 1.8 to 2.0 (260/280 nm) and > 2.0 for 260/230 nm . A volume of 20 ul per sample is sufficient.
The DNA samples have to be extracted using a CTAB-free protocol (best a spin column protocol), since very precise DNA sample quantification is critical for the success of the protocol.
The DNA samples have to be RNA-free. Thus, the DNA isolation protocol has to include an RNAse digestion step.
Before shipping us samples please email us gel images of representative samples. RR-Seq Sequencing is carried out with single-end 100 bp or single-end 150 bp reads.
We offer dual enzyme RR-Seq library sequencing with PstI or SbfI on one end combined with MspI for the other end.
Alternatively, we do offer a restriction-enzyme-free RR-seq protocol which is PCR-based.If there are no sequencing data for these enzymes for your species of interest yet, we would need to establish that the enzymes are suitable to avoid targeting sites present in abundant repeat sequences. We will do this by carrying out test-library preparations (see below).
Many types of RNA-seq require RNA samples of high integrity and high chemical purity – please see the sample requirements. If the tissue or cell samples are handled correctly (e.g. flash frozen and stored at -80C) standard spin column RNA extraction kits will yield RNA samples perfectly suitable for RNA-seq. Please note that samples destined for miRNA or small RNA studies need to be isolated with protocols specifically designed to retain the small molecules (please see below). Standard RNA isolation protocols will lead to the loss and sequence-specific selection of small RNA molecules. RNA samples should always be DNA-free. Nanodrop readings are more or less useless to determine RNA sample concentrations – please use fluorometric quantification instead (e.g. Qubit or Quantus instruments). The Nanodrop readings should be used to assess sample purity.
- For most tissues, the standard Qiagen RNeasy kits (cat. no. 74004) are perfectly fine (or similar kits from other vendors). These RNAeasy kits have to be used in conjunction with the Qiagen RNase-Free DNase kit (cat. no. 79254). We recommend to isolate the RNA, then perform the DNAse digestion in solution on the isolated RNA, and then clean up this reaction once more with the RNeasy kit. Alternatively, a kit like the Qiagen RNeasy Plus Micro Kit (cat. no. 74034) with “DNA Eliminator” gDNA removal columns may be used.
- In case the samples contain interfering chemicals or in case of very small sample amounts, it could be worth trying kits from Norgen Biotek. This manufacturer offers a selection of sample-type specific kits and uses a proprietary silicon carbide spin column matrix, which has a higher affinity for RNA compared to the standard silica columns. Thus, the Norgen Biotek kits often provide higher yields. Norgen kits offer two options for DNA removal: “plus” kits come with a dedicated DNA removal column; standard kits OTOH require DNAse treatment with the DNAse digestion add-on (Norgen cat. no. 25710).
- Always perform at least two spin column washes (with the kit wash buffer) after binding of the lysed sample to the column matrix.
- In case your lab uses a Trizol protocol for RNA isolations, we do recommend an additional sample purification with a spin column kit (e.g. Zymo RNA clean & concentrator 5 kit with DNAse – cat. no. R1013) for a DNAse digestion and clean up.
- An additional column cleanup is mandatory for RNA samples isolated from blood PAXgene or Tempus tubes (the tubes are used for blood preservation) after the initial RNA isolation. Often the preservative chemicals tend to contaminate the samples upon isolation. For the cleanup, we recommend the Zymo RNA clean & concentrator 5 kits with DNAse (cat. no. R1013) or similar.
- For miRNA and small RNA studies, protocols specifically designed to isolate also the shorter molecules have to be employed. Appropriate kits are available from multiple suppliers. Some suitable examples are an array of sample-type-specific NorgenBiotek and Qiagen kits as well as the Zymo Quick RNA kit. Please make sure that you apply the protocol variants designed to retain miRNAs for all of these.
- NEVER use heparin as an anticoagulant for blood samples destined for DNA or RNA sequencing.
- Avoid using glycogen (often recommended as co-precipitant).
- The 260/230 nm and 260/280 nm absorption ratio measurements (e.g. from NanoDrop) are should be used to assess sample purity. For RNA the 260/230nm ratio should be >1.5 and the 260/280nm ratio 1.8-2.1; Please see the sample requirements page and the sample cleanup FAQ.
How many cells will be needed to isolate sufficient RNA for conventional RNA-seq?
The typical mammalian cell contains 10 to 30 pg of RNA. Assuming the worst-case scenario (only 10 pg RNA content; 50% loss during isolation), you should be starting the RNA isolation with 20,000 or more cells to reach at least 100 ng total RNA sample, the lowest amount recommended for RNA-seq after poly-A enrichment. Please see the small sample RNA isolation recommendation above.
Avoiding Batch-Effects:
Both sample storage conditions and details of the RNA-isolation protocols are well-known to introduce technical variations into RNA-seq data. Because of this, it is recommended to:
- Isolate the RNA-samples in one batch.
- If RNA-isolations need to be carried out in several batches, they should be carried out by the same person using the same batch of reagents
- If RNA-isolations need to be carried out in several batches, the samples should be randomized between the RNA isolation batches (worth discussing with a statistician or the Bioinformatics Core).