
05 Sequencing
Depending on sequencer and in case of the HiSeq 4000 even depending on run type (single-end or paired-end) Illumina uses different approaches to sequence the indices. Please find detailed information here: indexed-sequencing-overview-guide-15057455-04-Illumina-pages1to8
The correct orientation of the barcode sequence fuehrer depends on the way the barcodes are added to the library. The gist of it is:
For barcodes added to Illumina libraries via a PCR step (e.g. Nextera; or also onto TruSeq stub-adapters):
- index 1 (i7) is always read as reverse complement of the sequence in TruSeq or Nexterastyle PCR oligos
- index 2 (i5) is read in direction of TruSeq or Nextera PCR oligos for Miseq, HS4000 SE (single-end), and NovaSeq
- index 2 (i5) is read as reverse complement of barcoded PCR oligos for NextSeq, iSeq, and HS4000 PE runs
For barcoded adapters added via ligation (e.g. standard Truseq style Y-adapters):
- index 1 (i7) is always read in direction(5’to 3′) of the sequence in TruSeq style oligo
- index 2 (i5) is read in direction of TruSeq or Nextera PCR oligos for Miseq, HS4000 SE (single-end), and NovaSeq
- index 2 (i5) is read as reverse complement of barcoded PCR oligos for NextSeq, iSeq, and HS4000 PE runs
Unfortunately, we cannot demultiplex inline barcodes.
Depending on sequencer model, the second index (called i5 index) will be read in different orientations. The correct adapter sequence information for the sample sheets is usually provided for both options by kit manufacturers. The two sequences are reverse complements of each other.
- The Forward Strand Workflow (previously known as Workflow A) sequences are required for MiSeq and AVITI.
- The Reverse Strand Workflow (previously Workflow B) sequences are required for NovaSeqs and NextSeq.
BTW, the AVITI demultiplexing software is smarter and can figure out the correct orientation on its own. Either orientation will be fine for AVITI submissions.
Here is the complete information on this topic from Illumina: https://support-docs.illumina.com/SHARE/IndexedSeq/indexed-sequencing.pdf
Certainly. Please provide a Bioanalyzer profile (we can also generate these), and barcode sequence information in the sample submission form. We will check the quantity of your libraries using real-time PCR (included in the sequencing price). We suggest to submit your library in at least 15 ul volume at a minimum concentration of 5 nM. Please see the Sample Requirements page for details. Depending on the sequencing platform, we can work with less library (down to 1 nM), but the quantification becomes less reproducible, the library becomes less stable, and relatively larger amounts of the library get lost sticking to the tube. The best buffer to store and submit libraries is 10 mM Tris/0.01% Tween-20 ph=8.0 or 8.4, but EB buffer is also acceptable. Please use 1.5 ml low-retention tubes ( e.g. Eppendorf DNA LoBind). If you do not provide a Bioanalyzer profile of your library, we will carry out the QC for a fee.
Please note that for the HiSeq3000/HiSeq4000 the libraries should have fragment lengths not longer than 550 bases and few molecules longer than 670 bp.
UMI is an acronym for Unique Molecular Identifier. UMIs are complex indices added to sequencing libraries before any PCR amplification steps, enabling the accurate bioinformatic identification of PCR duplicates.
UMIs are also known as “Molecular Barcodes” or “Random Barcodes”. The idea seems to have been first implemented in an iCLIP protocol (König et al. 2010).
UMIs are valuable tools for both quantitative sequencing applications (e.g. RNA-Seq, ChIP-Seq) and also for genomic variant detection, especially the detection of rare mutations. UMI sequence information in conjunction with alignment coordinates enables grouping of sequencing data into read families representing individual sample DNA or RNA fragments. Please see the graphic below.
The problems UMIs are addressing:
– Quantitative analysis: Many sequencing library preparation protocols enable high-throughput sequencing (HTS) from low amounts of starting material. Their preparation requires PCR amplification of the libraries. While the PCR polymerases and reagents have been improved greatly in recent years enabling a mostly unbiased amplification of sequencing libraries, some biases still remain against sequences with extreme GC contents and against long fragments. When starting from ultra-low input samples, stochastic effects in the first rounds of the PCR add to the problems. These issues can potentially cause erroneous quantitation data. Removal of PCR duplicates using alignment coordinate information is especially inefficient such for low input situations but also for deep sequencing data. In the latter case alignment coordinate-based de-duplification will remove large numbers of biological duplicate reads from the data, especially for the most abundant transcripts.
UMIs alleviate the PCR duplicate problem by adding unique molecular tags to the sequencing library molecules before amplification.
Please also see our FAQ: “Should I remove PCR duplicates from my RNA-seq data?” for more information.
– Rare variant analysis: Illumina sequencing provides data with low error rates (~0.1 to 0.5%) for most applications. These low error rates nevertheless interfere with the confident identification of low abundance variants. UMI-less data can’t distinguish between these and sequencing errors. UMIs in combination with deep sequencing yielding multiple reads for each of the sample DNA fragments solved this problem. The approach was first described as Duplex Sequencing. Hereby, single-strand consensus sequences (SSCSs) and Duplex consensus sequences (DCSs) assembly of the read families increase the accuracy of the sequencing data significantly. Please note that the DNA sample starting amounts and the library yields have to be controlled for this approach to be efficient. Applications include sequencing of heterogeneous tumor samples, cfDNA sequencing including ctDNA sequencing, deep exome sequencing.
The usage of UMIs is recommended primarily for three scenarios: very low input samples, very deep sequencing of RNA-seq libraries (> 80 million reads per sample), and the detection of ultra-low frequency mutations in DNA sequencing. For many other types of projects, UMIs will yield minor increases in the accuracy of the data. In addition, UMI analysis is an excellent QC tool of library complexity.
Incorporating UMIs into sequencing libraries:
– Our 3′-Tag-RNA-Seq protocols employ UMIs by default . For Tag-seq the first 6 bases of the forward read represent the UMI. These are followed by a common linker with the sequence “TATA”, followed by the 12 bp random priming sequence. It is recommended to transfer the UMI sequence information to the read header and to trim the first 22 bases from each read with UMI-TOOLS or custom scripts.
– For conventional RNA-seq and DNA sequencing applications you will specifically have to request UMIs on the submission form. The default library preparations will NOT use UMIs. The UMIs will be located in-line with the insert sequences for conventional RNA-seq, genomic DNA-sequencing, or ChIP-seq. The first twelve bases of both forward and reverse reads will represent UMIs and associated linker sequences (7 nt UMI sequence followed by a 5 nt spacer “TGACT”; UMIs of forward and reverse read are independent resulting in a combined UMI length of 14nt). UMIs and spacer are then followed by the biological insert sequences (for paired-end data a total of 22 bp will be dedicated to the UMIs instead of the inserts). The UMI and spacer sequences are usually trimmed off and the information transferred into the read ID header with software utilities like UMI-Tools or FASTP.
The figure below displays the (simplified) principles of the UMI data analysis for quantitative and variant detection studies.
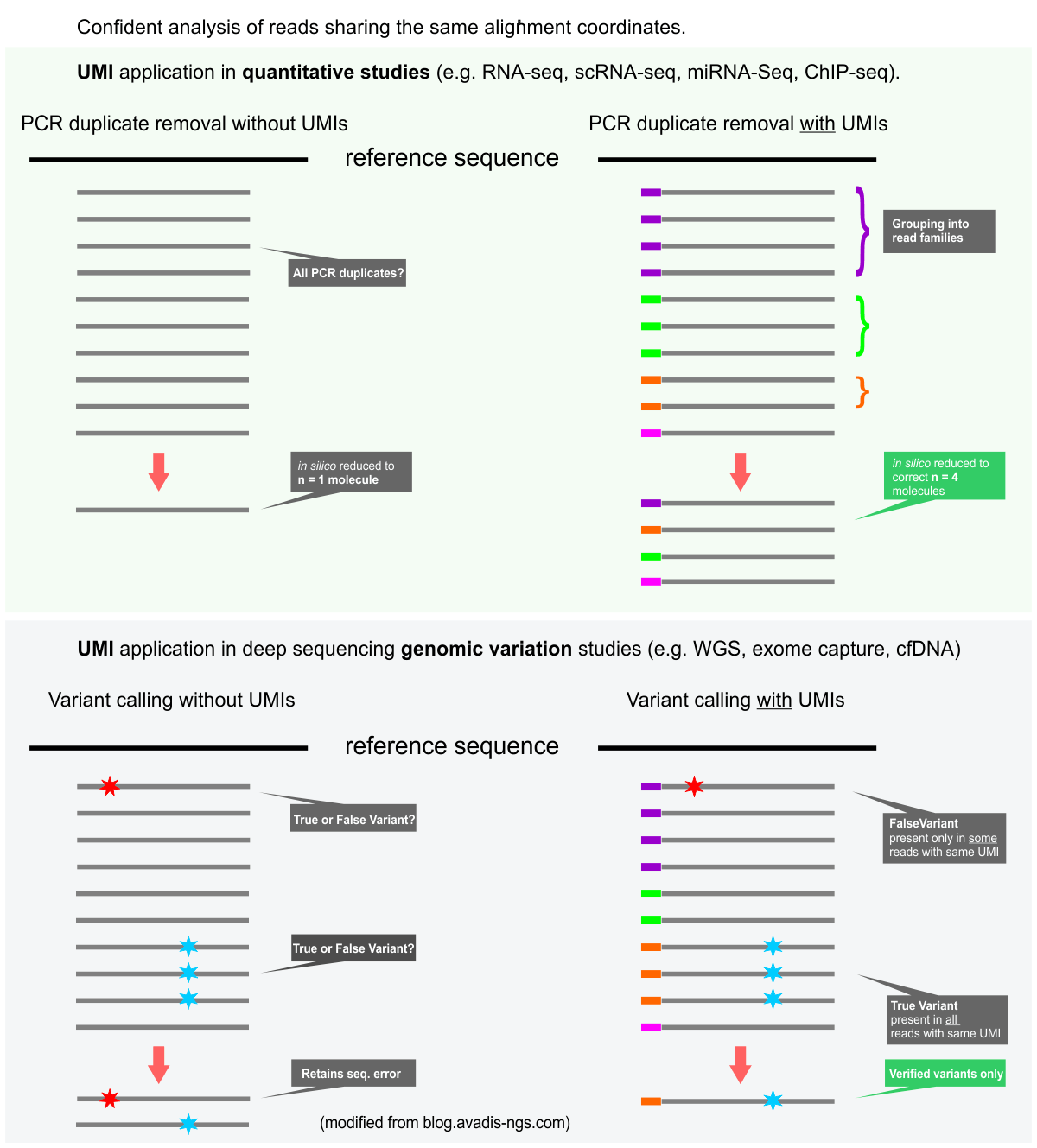
References:
Parekh et al 2016: The impact of amplification on differential expression analyses by RNA-seq. and
Fu et al. 2018: Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers.
Kennedy et al. 2015: Detecting ultralow-frequency mutations by Duplex Sequencing.
König et al. 2010: iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution.
Smith et al. 2017: UMI-tools: Modelling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy.
Software:
UMI-Tools: https://github.com/CGATOxford/UMI-tools
zUMIs: https://github.com/sdparekh/zUMIs
fastp: https://github.com/OpenGene/fastp (transfer of UMIs into read IDs)
Illumina has posted a Beginners Guide on their technology at:
https://www.illumina.com/science/technology/next-generation-sequencing/beginners.html
Please also see our information at: https://dnatech.genomecenter.ucdavis.edu/illumina-high-throughput-sequencing/
The Illumina specifications are based on the Illumina PhiX control library. Better or similar yields can be expected for other high complexity libraries (e.g. genomic, RNA-seq libraries) if they are within the recommended insert size ranges and do not average extreme GC-contents. Yields can vary depending on library type. For libraries that fulfill the criteria above, we do promise that the Hiseq 4000 and NextSeq sequencing data will exceed the Illumina yield specifications.
The table below displays the read numbers as CPF (clusters passing filter). For single-end sequencing CPF is equal to the read numbers. In case of paired-end sequencing the read number is twice the CPF figure.
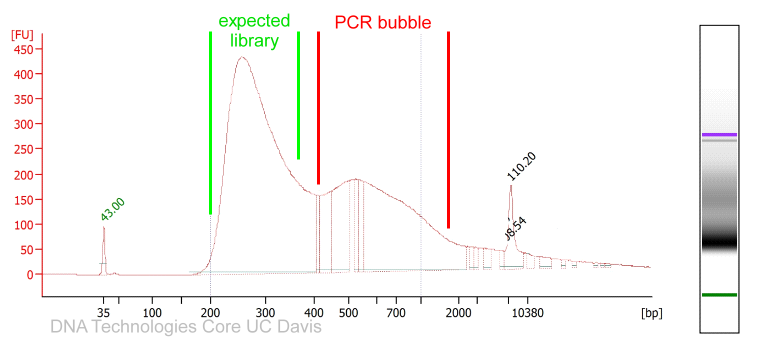
PCR amplified sequencing libraries frequently display library molecules seemingly about twice the excepted size or even bigger. In most cases, this phenomenon is caused by over-amplification of the libraries. These PCR artifacts do occur in cases the PCR reactions run out of essential reagents – in most cases the PCR primers will be exhausted. If primers are no longer available the PCR products will anneal to each other (the sequencing adapter sequence will be the by far most common sequences available). The resulting annealing products are often called “PCR-bubbles” and are partly double-stranded and partly single-stranded; thus they migrate considerably slower on agarose gels as well as on Bioanalyzer assays. Please see below.
Since these artifacts are merely annealing products, the resulting libraries are perfectly sequence-able. However, the quantification of such libraries by fluorometry will not be precise since the dyes used for these measurements are specific for double-stranded DNA molecules and PCR bubbles contain considerable amounts of single-stranded DNA that will not be measured. The PCR bubbles can be removed by amplifying the library one more time with a single cycle of PCR (a so-called “Reconditioning PCR“). For this PCR you could use standard Illumina P5 and P7 primers (please see below). The complementary sequences should be located at the very ends of all Illumina sequencing library molecules. In most cases PCR bubble artifacts can not be removed by SPRI bead size selections or Blue Pippin size selections; if necessary, a “Reconditioning PCR” is the best option.
However, to avoid unnecessary complexity loss of the library and introduction of polymerase errors, it would be best to optimize the library preparation protocol for a lower number of PCR cycles beforehand.
Reconditioning PCR protocol:
The reconditioning PCR uses standard Illumina P5 (5′-AATGATACGGCGACCACCGAGATCT-3′) and P7 (5′-CAAGCAGAAGACGGCATACGAGAT-3′) PCR primers which can be ordered as desalted DNA oligos.
Create a 10x concentrated primer mix at 20 μM each of these primers in EB buffer.
Add 1 to 4 μl previous PCR product, 2 μl 10x primer mix, 3 to 7 μl water for a total volume of 10 μl, then add 10 μl Kapa HiFi 2x Hotstart PCR master mix and pipette up and down several times.
Use the following cycling parameters: Initial denaturation 98°C 45 sec, denaturation 98°C 15 sec, annealing 60°C 30 sec, extension 72°C 30 sec, final extension 1 min.
Perform a standard Ampure XP/SPRI bead cleanup (e.g. with beads at 1.2x the sample volume) before analyzing the product by microcapillary electrophoresis.
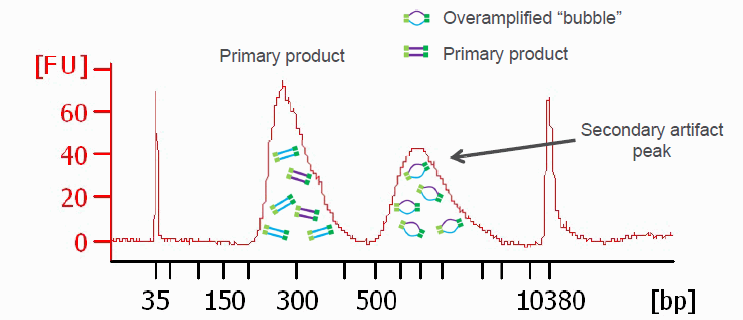
Another graphic illustrating PCR bubbles (source Illumina Inc.). Please also see: https://support.illumina.com/bulletins/2019/10/bubble-products-in-sequencing-libraries–causes–identification-.html
Illumina sequencers using the patterned flowcell technology (NovaSeq X, iSeq, NextSeq 2000, HiSeq 4000 &X Ten) can show an increased rate of barcode switching events. These artifacts are enabled by the exclusion amplification chemistry used on these sequencers; IIlumina calls the artifacts “index hopping”. Please see the index hopping information from Illumina here and in this video from Illumina. We have also have posted additional information here.
Please note that the AVITI sequencers, the MiSeq sequencer and NextSeq 500 chemistries are not sensitive to index hopping. For these sequencers single-indices and combinatorial indexing are fine.
To best avoid barcode switching read mis-assignments on new Illumina sequencers two measures are recommended:
- The use of uniquely dual-indexed (UDI) adapters. Illumina has collected information on the design of such adapters here. This adapter design employs unique barcodes for both index 1 and index 2 for each of the pooled libraries. Thus, 96 samples will use 96 different index 1 adapter sequences and 96 different index 2 adapter sequences.
- The efficient removal of any free primers and adapter-dimers from the libraries
The use of UDI adapters is highly recommended especially for the NovaSeq. Hereby, matching i5 and i7 indices per library must be avoided. UDI adapters can also be used on the MiSeq and the NextSeq, but they do not offer any significant advantages on these sequencers that employ the bridge-amplification chemistry. Further, any traces of free primers, primer-dimers, and adapter dimers should be removed from the sequencing libraries or the pools.
Commercial sources of UDI adapters: TruSeq-style uniquely indexed adapters are available from both Illumina and BiooScientific. Qiagen, NEB, and NuGEN are also supporting their library prep kits with optional UDI adapters. Nextera style indices need to be custom ordered from oligo vendors.
The DNA Technologies Core has 96-plex UDI adapter sets in stock that can be added to sequencing libraries by PCR. These barcode sets are available for both Nextera and TruSeq adapter designs. Please note that for TruSeq style libraries one will need to ligate a shortened and index-less stub-adapter instead of a standard Illumina adapter. The indices are then added after the cleanup of the ligation reaction by PCR.
Sequencing libraries prepared by the DNA Technologies Core:
The DNA Technologies Core uses UDI adapters for all library prep protocols that are compatible with dual indexing (e.g. DNA-Seq, RNA-seq, 3′-Tag-Seg, WGBS-Seq, …). The Core also makes sure to remove any traces of free primers, primer-dimers, and adapter-dimers.
- The fragment lengths should be consistent and best be between 100 and 300 bp (up to 400 bp for the majority of molecules is acceptable). Consistent fragment lengths can best be achieved on a Covaris style closed tube sonicator. We recommend avoiding probe sonicators.
- Please make sure to run the input controls on a Bioanalyzer or agarose gel beforehand, and email us an image of these.
- Sequence one “input control” per cell line/sample type.
- Analyze at least two biological replicates.
- We highly recommend verifying the enrichment of your regions of interest (e.g. promoter regions) vs. the control samples by qPCR, before submitting the samples for sequencing.
- For highest accuracy data we can now generate sequencing libraries with UMI-bearing sequencing adapters. UMIs (Unique Molecular Identifiers) allow the accurate detection and removal of PCR duplicate reads. This approach is especially recommended for low-input samples. The first nine bases of the forward and reverse reads will contain UMI sequences.
The required read number per sample will vary from target to target. For the study of point source transcription factors the ENCODE project recommends analyzing at least 20 million (uniquely mapping) reads (http://genome.cshlp.org/content/22/9/1813.long#boxed-text-2). Depending on the quality of your preps, perhaps 75% of the reads can be expected to be uniquely mapping. ENCODE tends to err on the high side with their recommendations. Thus, about 20 million read pairs per sample should be acceptable, but this is likely the minimum number.
Zhang et al. 2016 have studied the impact of the sequencing run types on ChIP-seq data analysis. Their data indicate that paired-end sequencing data provide significant advantages of single-end sequencing in ChIP-seq.
CUT&RUN sequencing might be a better alternative:
CUT&RUN sequencing (Skene & Henikoff 2017) is a faster protocol that for almost all applications is a more sensitive alternative requiring much lower cell numbers. CUT&RUN is suitable for studying histone modifications, transcription factors, and co-factors. In addition to lower input requirements CUT & RUN experiments also afford reduced read numbers (4 to 8 million read pairs per sample).
References:
Landt et al. 2012: ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Research 22: 1813-1831
Bailey et al. 2013: Practical Guidelines for the Comprehensive Analysis of ChIP-seq Data. PLOS Computational Biology https://doi.org/10.1371/journal.pcbi.1003326
Skene & Henikoff 2017: An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. https://doi.org/10.7554/eLife.21856
Zhang et al. 2016: Systematic evaluation of the impact of ChIP-seq read designs on genome coverage, peak identification, and allele-specific binding detection. BMC Bioinformatics volume 17, Article number: 96
MACS — Model-based Analysis of ChIP-Seq https://taoliu.github.io/MACS/
https://hbctraining.github.io/Intro-to-ChIPseq/lessons/05_peak_calling_macs.html
It is generally recommended to sequence 50 million or more reads/library-molecules per ATAC-seq sample for open chromatin detection and differential analysis (Buenrostro et al. 2015) and 200 million reads for TF footprinting (Yan et al. 2020). Preferred are paired-end sequencing data but single-end data are also usable. Most frequently ATAC-seq libraries are sequenced on the NextSeq with PE 75bp reads.
Paired-end data have slightly higher unique alignment rates, allow for PCR-duplicate removal and provide more complete information about the accessible sequences (due to the longer sequence information). Paired-end data allow for example the assignment of reads to categories such as nucleosome-free, mono-nucleosomal, and di-nucleosomal origins (Buenrostro et al. 2015) .
Controls are typically not run for ATAC-seq studies, but typically two biological replicates are required.
We generate libraries for sequencing with Illumina and PacBio instruments. Illumina libraries include genomic DNA, RNA-seq, ChIP-seq, micro RNA, small RNA, Methyl-seq, RRBS, and reduced representation libraries suitable for GBS analyses, Tag-Seq, and Tn-Seq. Please inquire with us if your protocol is not currently listed. Please see the Library Prep page and the Sample Requirements page for details. We carry out library QC via Bioanalyzer, library size-selections, pooling of sequencing libraries, and real-time quantitative PCR to accurately measure the concentration of sequence-able library molecules to achieve optimal sequencing output.
All sequencing data will be available for secure download via our SLIMS server.
Illumina sequencing data will be delivered as compressed FASTQ files. By default the data will be de-multiplexed (e.g. split according to sample). Each SLIMS directory will further contain a file with the de-multiplexing metrics and a file listing md5 checksums for all the FASTQ files. The checksums can be used to verify that the data integrity has been preserved your download/data transfer.
You will receive all the full-length reads passing the Illumina quality filter. Please contact us if you would like to receive the data quality-trimmed, adapter-trimmed, or adapter-filtered.
For PacBio sequencing you will receive all the data generated by the PacBio SMRT-Portal pipeline (raw data and primary analysis data) including FASTA and FASTQ files as well as the “bax.h5” and “.xml” files required to re-run the analyses. By default we are running also subread filtering as the secondary analysis (except for large gnome projects where this is not required) or other appropriate secondary analysis pipelines for bacterial genome assembly, amplicon analysis, or Iso-seq analyses. You will receive the complete secondary analysis data sets.
Please note: We do NOT archive sequencing data generated for you. Any sequencing data should be downloaded and verified as soon as possible.
The sequencing data will be available for download for three months after they are generated.
Work in the Core is performed on a “first come, first served” basis. HiSeq sequencing data will typically be delivered within about two to five weeks after library submission, while MiSeq has a 5-8 day turnaround time. The turnaround time can fluctuate and is dependent on the number of customer samples in the queue and the read types and lengths requested. Similarly library prep projects will be grouped according to the requested library prep protocols to allow for efficient processing.
Unfortunately not. The samples are put into library prep and sequencing queues after we have received both, the submission form and the sample. Submission forms have to be submitted in hard-copy (accompanying the samples) and electronically (please email the spreadsheet forms to us).
DNA samples and sequencing libraries will be stored at -20 degrees. RNA samples will be stored at -70 degrees C. However samples and libraries will only be stored for one year after sample submission. Please notify us two days in advance if you plan to pick up your samples. Please arrange the sample pick-up within two months of the data delivery. Please provide a FedEx account number and a shipping address if your samples should be shipped back. If possible submit aliquots of your samples to our cores for analysis and store a backup aliquot in your lab.
Please note that custom sequencing primers are not supported by Illumina — the company will not replace sequencing kits on run failures. Nevertheless, custom sequencing primers can enable some unique assays. It is certainly worth exploring if assays can’t be converted to use standard Illumina sequencing primers since standard primers are more or less guaranteed to work. Please also see the amplicon sequencing FAQ and note that the client carries the responsibility for any failures due to bad custom primer oligos or poorly designed custom primers.
On MiSeq sequencers custom sequencing primers can be used for three of the reads (forward, reverse, & 1st index reads)
On the NextSeq 550, custom sequencing primers can be used for all four reads (forward, reverse, & 1st and 2nd index reads)
Illumina does not support custom primers on the NovaSeqs at all.
For Illumina Sequencers: Please provide an aliquot for each custom primer (10 ul at 100 uM in EB buffer; low-bind tubes) with each library submission. Custom sequencing primers should be ordered HPLC purified to remove any incomplete oligos.
For the Element Bio AVITI sequencer: Please provide an aliquot for each custom primer (30 ul at 100 uM in low-TE buffer for index 1, index 2 and read 2 sequencing primers; 40 ul at 100 uM in low-TE buffer for read 1 sequencing primers ; low-bind tubes) with each library submission. Custom sequencing primers should be ordered HPLC purified to remove any incomplete oligos.
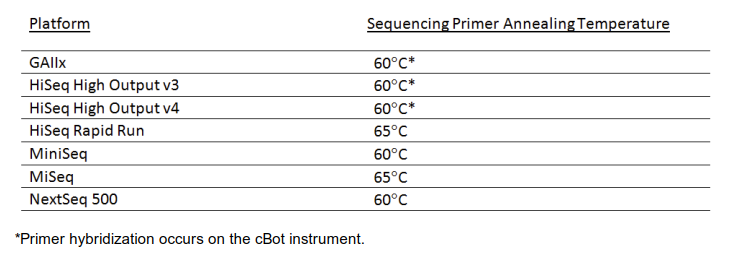
When designing custom sequencing primers, the melting temperature needs to be considered (among other criteria). It is suggested to calculate it with the IDT oligo analyzer for the default buffer conditions (50 mM Na+). Please see the recommended melting temperatures in the image below. While at the oligo analyzer site also check the designs for secondary structures, potential hairpin and self-dimers. In case the melting temperatures of your design are too low, the Tm can potentially be increased by inserting interspersed LNA bases into custom LNA oligos available from Qiagen. Avoid clustering of LNA bases and do not substitute the two last 3′ bases. LNA oligos are significantly more expensive than conventional oligos.
For more info please see this guide from Illumina: miseq-system-custom-primers-guide-15041638-01 and also the index read guide: indexed-sequencing-overview-guide-15057455-04-Illumina-pages1to8
Please note that the 2nd-index read is primed from a flowcell-bound oligo for the Miseq and most other Illumina sequencers. Thus, it cannot be customized. The sequencing of the second index begins with exactly seven dark cycles for which no sequence is recorded. Thus, the first base of the 2nd index sequence has to be the 30th nucleotide from the P5 end of the library molecule for most applications. Please note this FAQ can’t be a comprehensive guide to custom sequencing primer design and usage.
Small RNA-seq / miRNA-seq on the AVITI with libraries employing TruSeq Small RNA sequencing adapters:
Truseq Small RNA libraries will require custom sequencing primers on the AVITI. In most cases there will be no need for a reverse read, thus only TruSeq Small RNA Read 1 and i7/Index 1 custom sequencing primers will be required.
10x Specific recommendations: If using 10X Index Primer Set TS to complete their libraries (i.e. Single Cell Gene Expression Flex or Visium FFPE), they will need to spike-in a TruSeq Small RNA sequencing primer.
| Primer Name | Sequence (5′ -> 3′) | Tm (C) | Length (nt) |
| TruSeq Small RNA Read 1 | ATCTACACGTTCAGAGTTCTACAGTCCGACGATC | 65 | 34 |
| TruSeq Small RNA Read 2 | GTGACTGGAGTTCCTTGGCACCCGAGAATTCCA | 68 | 33 |
| i7/Index 1* | TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC | 68 | 33 |
| i5/Index 2 | Use Adept Index 2 Primer | — | — |
Common buffers for DNA samples, RNA samples, and primers are:
EB-Buffer: 10mM TRIS (pH= 8.0-8.4) – e.g. Qiagen EB Buffer
EBT-Buffer: 10mM TRIS, 0,1%Tween20 (pH=8.0-8.4)
TE-Buffer: 10mM TRIS, 1 mM EDTA (pH=8.0-8.4)
Low-TE buffer or TLE-Buffer: 10mM TRIS, 0.1 mM EDTA (pH=8.0-8.4)
All should be free of DNAses and RNAses.
The DNA Technologies and Expression Analysis Cores do not offer Sanger DNA Sequencing, but these services are available on campus. See the UC DNA Sequencing Facility or the CAES Genomics Facility for more information. There are multiple other service facilities that may be of interest, including the Veterinary Genetics Laboratory, CCM Mouse Biology Program, and Real-time PCR Research and Diagnostics Core Facility. If we missed out any UC Davis facilities that you would like us to list here, please contact us.