
01 General Information (20)
Please see this page: Getting Started. You will find information on how to set up an account, billing, data storage & distribution, scheduling, equipment & training, bulletins, research support, seed grants, and more.
The PPMS system is used to place orders (Core personnel only), to book shared instruments and schedule trainings.
Use of the PPMS system requires a one-time registration. The same system will be implemented in the near future by many other UCD Core labs.
We recommend submitting your PPMS account request as soon as possible since it will require a (fast) check/approval through our administration. The ‘group’ in PPMS is named after the PI, with other ‘users’ linked to the PI’s account – in general, it will be easier if the PI initiates the first PPMS account request.
Only customers (both the submitter and the PI) can enter financial/billing account information into PPMS; please make sure that the applicable financial account is entered before any submission.
Once the PPMS account is generated and the financial account is registered, please submit samples as described here. Upon arrival of the samples, the Core staff will enter the order in PPMS where you can review it. You will be notified by an email via PPMS.
To begin using PPMS:
- Ask your Principal Investigator to set up a lab group and a user account in PPMS here. On the menu bar, click on the Account Creation Request link. The group name has to be the name of the PI; the administrative/financial contact information is required; if possible a default KFS/financial billing account for the group should be entered; each user can add their billing account (or a PO for users outside the UC system) to the group. Non-UC customers wanting to pay by credit card, please leave the financial account information field blank. You will receive an invoice via email with instructions once your order has been processed.
- Then submit your personal account request.
- Wait to receive the PPMS account confirmation e-mail.
- Only customers can enter billing account information into PPMS! Before placing an order or submitting samples, please make sure that the desired KFS/financial billing account # is entered in PPMS (for users from other UCs the same applies to chart strings).
- For shared instrument users, notify the Core that your account has been created and let us know for which instruments you were trained on previously. The Core will then grant you user rights on the appropriate systems (instrument calendars) and you can begin making reservations in PPMS.
Please see these FAQ and PPMS account-setup help files for each of three user groups:
A general PPMS user help guide is available on the start page.
If you encounter any technical issues or have questions about PPMS, please see the PPMS FAQ, or email GCcoreadmin@ucdavis.edu .
Please also see our Getting-Started Guide with other administrative information on working with our Core and the Genome Center.
See this page for all the options: https://dnatech.genomecenter.ucdavis.edu/consultations/
Please pick a time & book your general consultation appointments with the Core director here: https://dnatechlutz.youcanbook.me/
The Genome Center Cores provide services at three recharge rate scales. The University of California rate scale (UC rates) applies to all projects paid through UC-system accounts. Non-profit/academic rates apply to all other non-profit research including government projects. The industry rates apply to all projects paid from for-profit businesses or institutions. Please make sure that you have a PPMS account to place requests and to enable invoicing.
Upon arrival of the samples, the Core staff will enter the order in PPMS where you can review it. You will be notified by an email via PPMS. You will be invoiced after you have received the data.
The invoicing process will vary slightly for UC Davis customers, other UC-system customers, and non-UC customers.
UC Davis customers:
- Once data are delivered to you, the project billing information will be entered into the secure PPMS system at https://ppms.us/ucdavis/login/?pf=5 , where you can view it.
- In the first days of a month your UC Davis DaFIS/KFS accounts will be billed processed.
Please note that the billing date (‘completed’) is recorded and that this is sufficient for expiring grants. The final processing can occur after the deadline (usually up to two months).
Other UC system customers:
- Once data are delivered to you, the project billing information will be entered
- into the secure PPMS system at https://ppms.us/ucdavis/login/?pf=5
- , where you can view it.
- In the middle of the following month the UC inter-campus transaction system will bill your account via an ITF.
Non UC-customers (academic, non-profit and industry):
- Once data are delivered to you, the project billing information will be entered into the secure PPMS system at https://ppms.us/ucdavis/login/?pf=5, where you can view it.
- Customers are sent an email notification within the first three days of the following month that lets them know their order is available to pay via credit card for 30 days. This email also requests customers notify our business administration: gccoreadmin@ucdavis.edu (530) 754-9648.
- Those that did not pay via credit card during the 30-day period are now manually invoiced and the credit card option in the Cores portal is turned off.
- The first days of the month are when invoices are processed by our business administration.
- Invoice customers generally receive their invoices after the 15th each month.
The UCD Genome Center accepts money transfers, credit card, and government purchase card (GPC) payments, etc.
Please find all the required information on sample submissions and project scheduling on this page http://dnatech.genomecenter.ucdavis.edu/sample-submission-scheduling/.
Non UC system users, both non-profit and for-profit organizations, can request invoicing (after data delivery) simply by entering the terms “credit card” or “invoice” in the account fields of the submission form. You can also set up a purchase order (a “PO”) beforehand and add the number to your PPMS account.
We require purchase orders only for institutions outside the US. Please email the POs to our Business Office (gccoreadmin@ucdavis.edu).
A purchase order is a simple letter from your financial administrators listing the requested services and prices.
It has to include the full contact information of your financial administrators as well as the name and institution of the Principal Investigator (“PI”).
The PO has to state the specific services requested from us – essentially repeating the information provided in quotes from us or from the recharge rate listing from our webpage.
POs should have an ID number on them which your administration can generate according to their preferences.
Please feel free to use your standard PO forms. This purchase order template, with all the required and suggested fields, is a suggestion: http://dnatech.genomecenter.ucdavis.edu/wp-content/uploads/2016/04/purchase-order-DNATech3_1.doc
Please see this page for more information on POs: http://blog.procurify.com/2013/09/23/all-you-ever-needed-to-know-about-purchase-orders/
We did modify a generic PO template from here: http://www.vertex42.com/ExcelTemplates/excel-purchase-order.html
Please see this page with the full contact information.
Lab questions:
Email: DNATECH@UCDAVIS.EDU
Lab phone: 530-754-9143
Business Office Administrator
–
Please see this FAQ: What Information Do You Require About My Project?
To be able to help you quickly with current or recent projects we always need the following information:
- PI name
- Submitter name
- Project ID (e.g. HiSeq3421)
- Submission date (even if vague)
Please contact Jen Stevens from our Business Office Administration for questions about contracts, bank transfers, credit card payments, DaFIS accounts, UC system account strings, and other payment options.
Katie Roberts
Business Office Administration
UC Davis Genome Center
451 Health Sciences Drive, 4303 GBSF
One Shields Avenue
Davis, CA 95616-5270
Ph: (530) 754-9648
gccoreadmin@ucdavis.edu
Please contact the core lab (dnatech@ucdavis.edu) for pricing questions, quotes, and simple billing questions or discuss your project with the core manager: https://dnatechlutz.youcanbook.me/
If you are interested in receiving information about new services, workshops and other updates from the DNA Technologies and Expression Analysis cores, please subscribe to the dnatech news lists.
Please always use the UC Davis email address, if applicable. We will send news of exclusively local interest to the dnatech_local_news list only and not try to not bother other subscribers with these. The UC Davis users will receive all news.
To subscribe to the lists, UC Davis users should send an email to sympa@ucdavis.edu with the following information in the subject line:
subscribe dnatech_local_news first_name last_name
–
To subscribe to the list, all non-UCDavis users should send an email to sympa@ucdavis.edu with the appropriate text in the subject line:
subscribe dnatech_news first_name last_name
Unsubscribe
You can unsubscribe from the list by simply sending an email to sympa@ucdavis.edu with the following information in the subject line:
unsubscribe dnatech_news
or
unsubscribe dnatech_local_news
Several pieces of equipment are available for use during normal working hours for a nominal ‘instrument use’ fee. To use the Shared Equipment, first you will need to be trained by Core staff (these is a training fee per person). Your PI will need to create an account with the Genome Center and supply a DaFIS # or PO for billing. Visit our login site and follow the instructions for “Creating an Account.” Also visit here to learn more about the Training and Use of Core Facility Equipment.
To schedule a time for instrument training, and to book instruments please use the PPMS system. Please see this page for PPMS information.
Consultations
We can provide better advice on appropriate or optimal sequencing options if you provide us important background information on your project. We suggest using the questions below as a starting point for inquiries.
Information requested for RNA-sequencing projects:
- Which species are you studying (prokaryotic or eukaryotic)?
- From which sample types will the RNA be isolated (e.g. tissues, cell cultures, whole organisms)?
- What is the objective of the study (e.g. differential gene-expression (DGE) study, gene annotation, identification of iso-forms)?
- For differential DGE studies: Are you interested only in gene-level DGE, or do you want to study the expression of the different transcripts of genes? Are you interested in non-coding RNAs?
- Do you expect lowly expressed genes to become important for your study?
- How closely are related (genetically) are the biological replicates?
- Do you expect to induce strong phenotypic changes by the varying treatments?
- What total RNA amounts do you expect to extract from the samples?
- What is the expected integrity of the RNA samples (e.g. RIN-scores)? Do you expect potentially interfering chemicals in the samples (e.g. phytochemicals)?
- How many samples do you want to study? Will the samples be available at the same time?
RNA-seq options are described on two pages: RNA-seq and 3′ Tag-Seq Gene Expression Profiling.
Information requested for DNA-sequencing projects:
- Which species are you studying (prokaryotic or eukaryotic)?
- What type of samples are you going to submit (e.g. genomic DNA, amplicons, metagenomic samples, purified strain or population samples)?
- What is the expected genome size? What genome coverage are you aiming for?
- What is the objective of the study (e.g. SNV variant analysis, genotyping, de novo genome assembly, structural genome variants, metagenomic analysis)?
- For de novo genome assemblies: Are you interested in genic regions or do you require the full genome sequence? Do you require the ordering of the assembly scaffolds?
- What DNA amounts do you expect to extract from the samples?
- What is the expected DNA sample integrity (fragment sizes)? Do you expect potentially interfering chemicals in the samples (e.g. phytochemicals)? What protocol are you using for the DNA isolation?
- How many samples do you want to study?
Most DNA-Seq options are described here.
In-Progress Project Inquiries
To be able to help you quickly with current or recent projects we always need the following information:
- PI name
- Submitter name
- Project ID (e.g. HiSeq3421)
- Submission date (even if vague)
You can use the map link below
Samples will be stored in the freezer for no longer than one year past the date of submission. DNA samples and sequencing libraries will be stored in a -20 degree C freezer. RNA samples are stored in a -80 C freezer. Please arrange for a pickup of your samples in the weeks immediately after data delivery.
We can also return samples to you via FedEx. This requires that we receive a PDF version of a completed shipping label from you using your FedEx account details. Please make sure to check dry-ice shipping for RNA samples when ordering the FedEx shipping label.
We are available every workday from 9 am to 5 pm.
We will be closed on university holidays. These are listed here: http://registrar.ucdavis.edu/calendar/holidays.cfm
This page informs on the data processing included with our sequencing services and bioinformatic analysis options.
This page informs on the data processing options for Infinium genotyping data.
Our neighbors from the Bioinformatics Core provide sequence data analysis, statistical evaluations, consulting, and training to help you get the most out of your data. Please contact us for joint consultations with the Bioinformatics Core staff and also for complete analysis packages including sequencing and bioinformatics (e.g. differential gene expression, variant calling).
Please support us by acknowledging our services in your publications. We have received NIH funding for the purchase of some of our instruments and this support should be mentioned.
Please add a sentence like this to your acknowledgments:
“The sequencing was carried out at the DNA Technologies and Expression Analysis Cores at the UC Davis Genome Center, supported by NIH Shared Instrumentation Grant 1S10OD010786-01.”
Acknowledgments like this are a big support for future NIH instrumentation grant applications. Thanks in advance!
Most of the technologies used in the Core are scientifically complex and expensive. It is expected that detailed conversations will take place as a matter of course between users and facility personnel at the outset and throughout projects. This does not warrant authorship but we love to see ourselves listed gratefully in the Acknowledgements section when your data is published. If extra effort is taken by us along the way, particularly in research where substantial intellectual and technical input is provided, we feel it is appropriate for the researcher to provide authorship to involved facility personnel. This is clearly a gray area in any scientific setting, and because fees are involved it’s even grayer. Just remember that the cheerful professionals that help you out also have resumes, come up for performance reviews, and citations are important additions to their CV. We will initiate discussions on this early in a project if we feel it’s relevant. So far everyone has been more than accommodating, so thank you all for that!
The Genome Center invites proposals for pilot projects that merit support by the Genome Center. These seed grants award up to $2,000 to spend at any of the Cores. Only UC Davis labs can be eligible for these grants.
The Genome Center seed grants are available year-round. For the DNA Technologies Core, the seed grant program has two major objectives:
- introduce UC Davis labs to high-throughput sequencing or genotyping methods (applies to labs that have not used our technologies before)
- support the development of new genomic and transcriptomic methods/protocols
A seed grant application should be informal but should be about one page long and include:
- a short introduction on the scientific question being addressed and a short outline of the project
- a sentence on how the project would fit in with at least one of the objectives of the seed grant program
- an outline of the planned data analysis (will there be a qualified analyst and computational resources available ?)
- a listing of the specific services to be provided by our Core
- a budget (including an account number in case the costs exceed the grant amount).
We highly suggest discussing your proposal prior to submission of an application with the core manager. Please see the consultation link on the home page.
After approval of a seed grant, samples have to be submitted within three months, for the project to stay eligible for the seed grant support. Please understand that funds will be allocated to other projects, if the time frame is exceeded.
Equivalent pilot grants are also available through the Bioinformatics core. Please contact the Bioinformatics Core here: bioinformatics.core@ucdavis.edu.
The BGI@UCDavis was located in the School of Medicine on the Sacramento campus and did provide sequencing services for large scale sequencing projects up until September 2015. This BGI facility is now closed. Since our DNA Technologies Core operates the latest generation of sequencers (HiSeq3000/4000), which offers a 7 times increased throughput compared to the previous generation, our Core is now taking on also large sequencing projects. The DNA Technologies Core is offering all the services previously provided by the BGI@UCDavis. In addition we offer many specialized services and support for custom sequencing projects.
02 Prices or Recharge Rates (4)
Please check our Prices page for the complete list of pricing for genotyping and sequencing.
The Genome Center Cores provide services at three recharge rate scales. The University of California rate scale (UC rates) do apply to all projects paid through UC-system accounts. Non-profit/academic rates do apply to all other non-profit research including government projects. The industry rates apply to all projects paid from for-profit businesses or institutions.
The Genome Center Cores provide services at three recharge rate scales. The University of California rate scale (UC rates) applies to all projects paid through UC-system accounts. Non-profit/academic rates apply to all other non-profit research including government projects. The industry rates apply to all projects paid from for-profit businesses or institutions. Please make sure that you have a PPMS account to place requests and to enable invoicing.
Upon arrival of the samples, the Core staff will enter the order in PPMS where you can review it. You will be notified by an email via PPMS. You will be invoiced after you have received the data.
The invoicing process will vary slightly for UC Davis customers, other UC-system customers, and non-UC customers.
UC Davis customers:
- Once data are delivered to you, the project billing information will be entered into the secure PPMS system at https://ppms.us/ucdavis/login/?pf=5 , where you can view it.
- In the first days of a month your UC Davis DaFIS/KFS accounts will be billed processed.
Please note that the billing date (‘completed’) is recorded and that this is sufficient for expiring grants. The final processing can occur after the deadline (usually up to two months).
Other UC system customers:
- Once data are delivered to you, the project billing information will be entered
- into the secure PPMS system at https://ppms.us/ucdavis/login/?pf=5
- , where you can view it.
- In the middle of the following month the UC inter-campus transaction system will bill your account via an ITF.
Non UC-customers (academic, non-profit and industry):
- Once data are delivered to you, the project billing information will be entered into the secure PPMS system at https://ppms.us/ucdavis/login/?pf=5, where you can view it.
- Customers are sent an email notification within the first three days of the following month that lets them know their order is available to pay via credit card for 30 days. This email also requests customers notify our business administration: gccoreadmin@ucdavis.edu (530) 754-9648.
- Those that did not pay via credit card during the 30-day period are now manually invoiced and the credit card option in the Cores portal is turned off.
- The first days of the month are when invoices are processed by our business administration.
- Invoice customers generally receive their invoices after the 15th each month.
The UCD Genome Center accepts money transfers, credit card, and government purchase card (GPC) payments, etc.
The Genome Center Cores (including the DNA Technologies Core) provide services at three recharge rate scales.
- The University of California rate scale (UC rates) do apply to all projects paid through UC-system accounts.
- Non-profit/academic rates do apply to all other non-profit research including government projects.
- The industry rates apply to all projects paid from for-profit businesses or institutions.
Please note that affiliations with universities are not a determining factor with regard to which of the recharge rate scales applies.
Non UC system users, both non-profit and for-profit organizations, can request invoicing (after data delivery) simply by entering the terms “credit card” or “invoice” in the account fields of the submission form. You can also set up a purchase order (a “PO”) beforehand and add the number to your PPMS account.
We require purchase orders only for institutions outside the US. Please email the POs to our Business Office (gccoreadmin@ucdavis.edu).
A purchase order is a simple letter from your financial administrators listing the requested services and prices.
It has to include the full contact information of your financial administrators as well as the name and institution of the Principal Investigator (“PI”).
The PO has to state the specific services requested from us – essentially repeating the information provided in quotes from us or from the recharge rate listing from our webpage.
POs should have an ID number on them which your administration can generate according to their preferences.
Please feel free to use your standard PO forms. This purchase order template, with all the required and suggested fields, is a suggestion: http://dnatech.genomecenter.ucdavis.edu/wp-content/uploads/2016/04/purchase-order-DNATech3_1.doc
Please see this page for more information on POs: http://blog.procurify.com/2013/09/23/all-you-ever-needed-to-know-about-purchase-orders/
We did modify a generic PO template from here: http://www.vertex42.com/ExcelTemplates/excel-purchase-order.html
03 Sample Preparation & Sample Requirements (20)
DNA Sample Integrity:
For Illumina short-read sequencing:
DNA sample integrity should best be QC-ed by agarose gel-electrophoresis and ethidium bromide staining. “Safe” gel-stains such as Gel-Red work just as well.
These stains will make both DNA and RNA visible. RNA will run as an halo-like smear in the range 50 to 300 bp.
We suggest a 1% agarose gel and a ladder marker that best includes a 20 kb band like the GeneRuler 1kb Plus DNA ladder from Thermo Scientific. Please load about 40 to 100 ng DNA for each sample. Other conditions can work as well.
The agarose gel image will show the presence or absence of RNA contamination and provide the best information on potential sample degradation.
Please email us an agarose-gel image before shipping the samples in case of any concerns. Please always ship a copy of the agarose-gel image together with the samples.
For PacBio or Nanopore long-read sequencing:
HMW-DNA samples should be QC-ed via pulsed-field gel electrophoresis (PFGE) or field-inversion gel electrophoresis (FIGE). We can carry out this QC for you. The Femto-Pulse will instrument enables capillary FIGE with ultra-low input amounts and will provide a digitized data analysis (similar to the Bioanalyzer for short molecules).
If you do not have access to these technologies, we suggest running a longer conventional agarose gel (as described above) to get a first idea about the sample quality before shipping the samples to us for a FIGE analysis.
Please always ship a copy of agarose-gel images together with the samples.
DNA Sample Purity:
DNA sample purity has to be determined via spectrometry. Please see the sample requirements page for the recommended values for your protocol. It is certainly helpful to also record the entire UV absorption spectrum as it provides additional information. For DNA samples the 260/230nm ratio should be >2 and the 260/280nm ratio 1.8-2.0 .
Please also see:
Which DNA isolation protocols do you recommend for Illumina sequencing?
How should I purify my samples? How should I remove DNA or RNA contamination?
Do you offer DNA isolations and RNA isolations as a service?
How do I prepare DNA samples for RR-Seq (reduced representation sequencing)?
- Spin column DNA isolation kits are available from multiple vendors including Qiagen, Zymo, Omega Biotek, Sigma, and Norgen Biotek e.g. Qiagen DNeasy Blood & Tissue kit with added RNAse A (RNase A 100 mg/ml; cat. no. 19101).
- The Qiagen DNeasy Blood & Tissue kit (with added RNAse A) is also the default kit for bacterial isolate DNA extractions. The kit comes with dedicated bacterial protocols.
- Some vendors also offer DNA isolation kits in a 96-well spin-plate format for large sample numbers (e.g. Qiagen, Zymo).
- Only use a protocol that includes an RNase digestion step to remove any contaminating RNA; RNA can inhibit the DNA sequencing library preparation.
- Plant samples will require a dedicated kit that includes a lysis buffer designed to capture harmful plant chemicals like phenols (e.g. Qiagen DNeasy Plant). Without protective additives in the lysis buffers, plant chemicals will damage the DNA.
- Similarly, soil samples are rich in inhibitors of enzymatic reactions. Dedicated protocols and kits that can remove such chemicals (e.g. DNeasy Powersoil Pro) and are highly recommended.
- If accurate quantification of the resulting DNA samples is required, absolutely avoid any protocols that employ the chemical CTAB. Spin column protocols are usually CTAB-free.
- To achieve the cleanest DNA isolation, only use at most half the sample amount of the maximum recommended by the manufacturer.
- Spin-column isolation tips: perform the “optional” steps described in the manufacturers manual. Always perform at least two spin column washes (with the kit wash buffer) after binding of the sample to the column matrix. Add a short “dry spin” of the column after the washes and before the elution buffer addition to avoid any carryover of the ethanol wash buffer. Extend the incubation times for elution of DNA samples from spin columns to at least 5 minutes – or perform two consecutive elutions instead.
- NEVER use heparin as an anticoagulant for blood samples destined for DNA or RNA sequencing. EDTA (preferred) or citrate anticoagulants should be used. Heparin co-purifies with nucleic acids and inhibits multiple types of enzymes like polymerases and ligases.
DNA Sample QC:
- After extraction the DNA sample purity has to be determined via spectrometry (e.g. Nanodrop). Please see the sample requirements page for the recommended values for your protocol. It is certainly helpful to also record the entire UV absorption spectrum as it provides additional information. For DNA samples the 260/230 nm ratio should be >2 and the 260/280 nm ratio 1.8-2.0.
- To assess the DNA sample integrity and verify the removal of RNA, DNA samples should best be analyzed by agarose gel electrophoresis, see: How should I QC my genomic DNA samples before sequencing? Please email us an agarose gel electrophoresis image with the DNA samples. For spin-column protocols the DNA fragments should be longer than 10 kb or 15 kb. Shorter fragments indicate DNA damage before the DNA isolation; please inquire with us in such cases.
Please also see:
How should I QC my genomic DNA samples before sequencing?
How should I purify my samples? How should I remove DNA or RNA contamination?
Do you offer DNA isolations and RNA isolations as a service?
How do I prepare DNA samples for RR-Seq (reduced representation sequencing)?
Assessing the integrity of RNA samples with the Bioanalyzer can be time-consuming and expensive since each run takes an hour and only 12 RNA can samples can be run.
To make RNA QC more convenient and affordable we will be running one or more batches of RNA samples weekly on the high-throughput LabChip GX.
For UC Davis labs the cost per sample will be $5 (with a minimum of $10).
In order to generate usable data:
- Provide sample names with an implicit order. The plots of the traces will identify the well position not sample names.
- We will not adjust sample concentration or volumes. It is your responsibility to meet the sample requirements.
- Submit total RNA samples in a well-sealed 96-well plate or in strip tubes (please see this page for examples) and a filled out QC submission form.
- For large sample numbers make sure that the plates are clearly labeled. Please use the QC submission form simply as a cover sheet in this case, listing the plate names and the number of samples for each. Samples should be filled into plates in column order from A1 to H1, then A2 to H2, etc. …
- Each RNA sample needs to have a volume 2 ul to 6 ul and contain 30 ng to 250 ng total RNA (this is the amount, not the concentration).
- Glycogen can interfere with the RNA QC and should be avoided (it can interfere with both spectrometry as well as capillary electrophoresis on the LabChip and the Bioanalyzer).
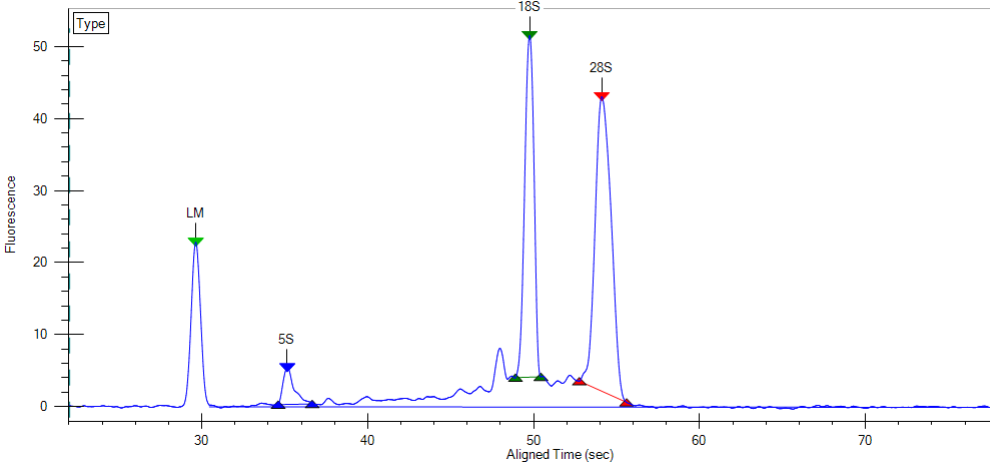
The LabChip GX will generate traces like the one below and RNA Quality Scores which are similar to the RIN scores provided by the Agilent Bioanalyzer and can be used interchangeably.
Please note that the capillary electrophoresis will be of lower resolution compared to the Bioanalyzer, but “good enough”. There is also no dedicated quality score algorithm for plant samples on the LabChip. The scores it produces are still realistic.
Unfortunately, we cannot demultiplex inline barcodes.
Depending on sequencer model, the second index (called i5 index) will be read in different orientations. The correct adapter sequence information for the sample sheets is usually provided for both options by kit manufacturers. The two sequences are reverse complements of each other.
- The Forward Strand Workflow (previously known as Workflow A) sequences are required for MiSeq and AVITI.
- The Reverse Strand Workflow (previously Workflow B) sequences are required for NovaSeqs and NextSeq.
BTW, the AVITI demultiplexing software is smarter and can figure out the correct orientation on its own. Either orientation will be fine for AVITI submissions.
Here is the complete information on this topic from Illumina: https://support-docs.illumina.com/SHARE/IndexedSeq/indexed-sequencing.pdf
To check if the genome of your species of interest is suitable for Optical Genome Mapping on the Bioanao Saphyr, you should check the distribution of labeling sequence motifs. For this purpose, Bionano provides in silico digestion tools with the “Label Density Calculator” program. “Bionano Access” also has such a feature. Both programs are available from this webpage: https://bionanogenomics.com/support/software-downloads/
Quality and quantity of DNA and RNA is critical for high quality sequencing output. Please make sure your DNA is not degraded and is free of RNA contamination. RNA samples should always be assessed on the bioanalyzer for the absence of gDNA contamination (can be removed with DNaseI treatment followed by a column clean-up; e.g. Zymo “RNA Clean and Concentrator”) and degradation. Preferentially determine the concentrations of your DNA and RNA samples using fluorometry (e.g. with a Qubit or plate reader). The sample purity should be assessed by spectrophotometry (e.g. Nanodrop). Please see this page for a comprehensive table of sample requirements for sample QC, library preps, or your self-made libraries. Please see the Library Prep Page for details on the library prep processes. For submission information, including submission forms and shipping details, please visit the Sample Submission & Scheduling page. If you are submitting DNA for PacBio libraries, please follow the PacBio Guidelines for Shipping and Handling.
The Real-time PCR core can carry out DNA as well as RNA extractions for you.
What type of samples are recommended for the isolation of HMW-DNA? (for Long-Read Sequencing)
Please see the information in this PDF that we wrote originally for the California Conservation Genomics Program (CCGP).
It contains recommendations for the collection of samples for both DNA and RNA isolations for the purpose of reference-quality genome assemblies and gene annotations.
Sample Collection Recommendations for Long-Read Sequencing and Gene Annotations
What type of samples are recommended for RNA isolations for gene annotations?
Please see the information on page three of this PDF that we wrote originally for the California Conservation Genomics Program (CCGP).
It contains recommendations for the collection of samples for both DNA and RNA isolations for the purpose of reference-quality genome assemblies and gene annotations.
Sample Collection Recommendations for Long-Read Sequencing and Gene Annotations
The sample amount requirements are chosen to ensure both, high-quality data and efficient processing.
Most of the library prep protocols will generate sequenceable libraries with lower input amounts than request, often requiring additional PCR cycles.
Processing often low-input samples often requires additional handling and QC steps. Thus, additional custom processing costs may apply.
Please contact us before submitting such samples. When working with sample amounts lower than recommended, you will generally run the risk of introducing biases and noise into your data. This may or may not be an acceptable trade-off for your specific project.
The sample integrity requirements are chosen to ensure the generation of high-quality data. Please contact us before submitting such samples. It may be possible to use an alternative protocol that tolerates some sample degradation.
When working with samples of lower integrity than recommended, you will generally run the risk of introducing biases and noise into your data. This may or may not be an acceptable trade-off for your specific project.
We can certainly work with degraded samples if requested, but we cannot vouch for the quality of the resulting data.
Processing of low-integrity samples often requires additional handling and QC steps. Thus, additional custom processing costs may apply.
Bead based sample cleanups (e.g., Ampure XP, RNAClean XP) and spin column-based protocols (e.g., Qiagen, Zymo, NorgenBiotek) tend to be the most efficient ways to remove chemical contaminants. For genomic DNA samples to be sequenced on Illumina sequencers, we suggest spin columns since DNA treated this way will always dissolve well and completely.
Please test for chemical contamination by spectrophotometry (e.g., Nanodrop), concentrations should be measured by fluorometry instead (Qubit, Quantus, plate reader, …) :
- Please see this guide from the University of Arizona on the interpretation of Nanodrop data. Skewed absorption ratios indicate that there is chemical contamination, but not precisely which contaminant and if it will be deleterious or not,
- The 260/230 nm and 260/280 nm absorption ratio measurements are most frequently used to assess purity. Please see the sample requirements page for the recommended values for your protocol. However, it is certainly helpful to also record the entire UV absorption spectrum as it provides additional information. For RNA the 260/230nm ratio should be >1.5 and the 260/280nm ratio 1.8-2.1; For DNA the 260/230nm ratio should be >2 and the 260/280nm ratio 1.8-2.0 .
- In case the absorption ratios are skewed, it is often worth checking if any alcohol was carried over from the spin column or bead washes. Any organic substance, including ethanol, will skew the 260/230 nm ratios. One can vent the open sample tube (for example for 20 minutes) on the lab bench and measure again afterwards to see if the contamination has disappeared.
- The spectrophotometer ratios themselves become easily misleading at very low DNA or RNA concentrations (10 ng/ul or less). In these cases the nucleic acid samples contribute very little to the signal and the slightest contamination dominates the readings. Please record the absorption spectra.
Multiple protocols are available to remove DNA or RNA contaminants. Please find our suggestions for affordable solutions for Illumina sequencing below.
RNA samples need to be DNA-free. The RNA isolation protocol should always include a DNase digestion step; in problematic cases use RNA-clean & concentrator kits with DNase. On an agarose gel, DNA contamination will be visible as a smear or band of fragments considerably larger than the RNA (>10 kb). On Bioanalyzer RNA-chips, DNA contamination will be visible in the size range 4 kb to 10 kb.
If you are using a Trizol protocol for the RNA extractions we would highly recommend cleaning the samples afterwards with a spin column kit (e.g. RNA-clean & concentrator kits) to remove any phenol traces.
Please note that the additional column cleanup is mandatory for RNA samples isolated from blood PAXgene or Tempus tubes (for blood sample preservation) or with the accompanying PAXgene and Tempus RNA isolation kits.
DNA samples need to be RNA-free. The DNA isolation protocol should always include an RNase digestion step; in problematic cases we recommend using RNase I (e.g. add 1 ul RNAse I to your sample and incubate at 30 degrees C for 20 minutes). RNase I does not require a special buffer (it works in TE buffer). For the removal of the RNase I, Ampure XP beads (or similar) or DNA-clean & concentrator kits will work fine (we suggest extending incubation times for elutions from the columns to at least 5 minutes or to perform two elutions). Do NOT try to inactivate the RNAse by heating (the NEB manual suggests heating to 70°C – this will already denature DNA dissolved in water or EB buffer and introduce biases in the library preparation!
DNA samples can be QC-ed easily by agarose gel electrophoresis and ethidium bromide staining. The stain will make both DNA and RNA visible. RNA will run as an halo-like smear in the range 50 to 200 bp.
For the removal of chemical contaminants solid-phase paramagnetic bead cleanups (SPRI-beads) are a solution suitable for high-throughput processing. The first such products were Ampure XP (for DNA) and RNAClean XP from Agencourt/Beckman. Many companies are now selling lower-cost versions, for example MagBioGenomics DNA beads and RNA beads.
We can recommend this EdgeBio magnetic plate for bead cleanups in 96-well plates.
- For spin-column cleanups: Please perform the optional steps described in the manual. Always perform at least two spin column washes (with the kit wash buffer) after binding of the sample to the column matrix. Also, add a short “dry spin” of the column after the washes and before the elution buffer addition to avoid any carryover of the ethanol wash buffer.
- We suggest extending incubation times for elutions of DNA samples from spin columns to at least 5 minutes – or to perform two consecutive elutions instead.
- NEVER use heparin as an anticoagulant for blood samples destined for DNA or RNA sequencing. EDTA (preferred) or citrate anticoagulants should be used. Heparin co-purifies with nucleic acids and inhibits multiple types of enzymes like polymerases and ligases.
- Avoid using glycogen as co-precipitant.
DNA samples for long-read sequencing library preparations or also 10X genomics linked-reads have to be exceptionally pure. Please see the sample requirements. For difficult DNA samples, especially all plant DNA samples with hard-to-remove contaminants (e.g. some polysaccharides), we recommend to carry out a high-salt/phenol/chloroform cleanup (please see this protocol) . Please note that this protocol often leads to a loss of 50% of the sample. Alternatively the the BorealGenomics Aurora instrument (discontinued but still available, please inquire) can be used. This process is slower though (only a single sample can be processed per day or two days) and is accompanied by similar or greater samples loss.
At the moment we only carry out high-molecular-weight DNA (HMW-DNA) isolations for the purpose of 10X Genomics and Nanopore sequencing. Please inquire with Ruta Sahasrabudhe, PhD.
We do not offer DNA isolations for Illumina sequencing and RNA isolations at the moment. However the Taqman Core does. Please contact the Real-time PCR and Research Diagnostics Core (also known as the Taqman Core). They carry out nucleic acid isolations from a wide variety of tissues for both plant, animal, bacterial , and fungal samples. The Taqman Core manager Samantha Barnum and her team have many years of experience in the extraction of sequencing-worthy DNA and total RNA samples. Please note that for sequencing purposes the ‘Qiagen Nucleic Acid Extraction’ option should be selected – this protocol generates the highest quality material for Illumina sequencing for both DNA and RNA. The same protocol may also be suitable for PacBio sequencing, but is only recommended for bacterial samples.
We encourage scheduling your DNA and RNA extraction services directly with the Taqman Core and to mention that the samples are designated for sequencing. Please contact the Taqman Core manager Samatha Barnum for technical details/sample requirements.
There are multiple valid protocols available for amplicon sequencing on Illumina systems. Here we describe one of many options: A two-step PCR protocol to generate complete sequencing libraries.
This protocol has the advantage that it does not require custom sequencing primers and that the barcode-indexing oligos can be re-used for multiple different amplicons and future projects. We suggest to follow a “16S amplicon” protocol that was explicitly designed by Illumina to be adaptable to other targets (please see the full protocol and pages 3 and 4 here).
Once you have designed the oligos as described in the Illumina protocol (forward overhang plus your sequence-specific primer as well as reverse overhang plus sequence-specific primer), we suggest checking these sequences on the IDT oligoanalyzer ( https://www.idtdna.com/calc/analyzer ) for secondary structures. It is advisable to avoid any sequences that generate a Delta G smaller than -9 for any of the structures.
There is no need to purchase an Illumina Nextera index kit. The sequences for the index primers (26 i7 index 1 sequences; 18 i5 index 2 sequences) are available on pages 7 and 8 here. These indices allow for the combinatorial sequencing of up to 468 samples. When ordering oligos please use the index sequences in the “Bases in Adapter” columns. The oligos are used for standard PCR reactions. Thus, low-cost desalted oligos can be ordered for this purpose anywhere and will work just fine. We strongly recommend using plates with single-reaction aliquots of these index primers for your experiments to make sure that index primer stocks cannot become contaminated.
Your first round PCR amplicon products will have universal tails/tags/overhangs on both ends. Since you can use dual indexes, you could order for example 5 index oligos with i5 indexes and 5 index oligos with i7 indexes and have 25 usable barcode combinations for your project. If you are using single indices they have to be i7 (P7 adapter) indices. However, for HiSeq 4000 and NovaSeq sequencing you should use uniquely-dual-indexed (UDI) barcode combinations.
The first round PCR primer designs use Nextera-style tag sequences (overhang sequences) and look like this:
Forward overhang P5-tag: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-[locus-specific sequence]
Reverse overhang P7-tag: 5’ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-[locus-specific sequence]
The second round PCR primers are Nextera-style index primers – i5 and i7 indicate the location of the barcode index sequences:
P5-PCR index primer: 5’ AATGATACGGCGACCACCGAGATCTACAC[i5]TCGTCGGCAGCGTC
P7-PCR index primer: 5’ CAAGCAGAAGACGGCATACGAGAT[i7]GTCTCGTGGGCTCGG
Please optimize the conditions of the first round PCR to avoid primer-dimer generation. The PCR reactions should be cleaned up with Ampure XP beads (or similar) and resuspended in EB buffer.
Once you have verified (via agarose gel electrophoresis) that the PCR products for all samples are clean and of about the same and expected size, the samples should be pooled equimolarly. We suggest to quantify the samples via fluorometry (Qubit or plate reader) for accurate pooling.
In case you are targeting only a single amplicon, it helps to create sequence diversity by adding a set of PCR primers with added diversity spacer “N” bases (or defined bases; up to seven of them) between the overhangs for both forward and reverse primers (Fadrosh et al. 2014, Wu et al. 2015). The resulting set of primers should be pooled in equimolar ratios and used for the first round of PCR.
The original Illumina design looks like this: overhang+locus-spec. sequence (no spacer):
5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐[locus‐specific sequence]
Complementary stagged spacer versions of this oligo would be:
One spacer base added: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐X-[locus‐specific sequence]
Two spacer bases added: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐XX-[locus‐specific sequence]
Three spacer bases added: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐XXX-[locus‐specific sequence]
Four spacer bases added: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐XXXX-[locus‐specific sequence]
Five spacer bases added: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐XXXXX-[locus‐specific sequence]
Six spacer bases added: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐XXXXXX-[locus‐specific sequence]
Seven spacer bases added: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐XXXXXXX-[locus‐specific sequence]
Knowing the locus-specific sequence one can certainly be smarter and make the two “N”s directly before the locus‐specific sequence different from the first two bases of the locus‐specific sequence (Fadrosh et al. 2014). If pooling amplicons for multiple targets (more than 8) there is no advantage using diversity spacers.
Some downstream programs might require the removal of the diversity spacers. dbcAmplicons can demultiplex the data as well as trim/remove the diversity spacer.
Fungal ITS: Illumina has published a second version of this protocol, modified to sequence and study fungal ITS sequences.
Qiagen offers a commercial amplicon prep kit for multiple 16S regions and ITS for which they have perfected the diversity spacer approach described above. This kit eliminates the need for PhiX spike-ins.
A much more detailed protocol for 16S and other amplicon sequencing is available here: Gohl et al. 2016
Please see this page for the library requirements for sequencing (http://dnatech.genomecenter.ucdavis.edu/sample-requirements/). The above protocol will generate a surplus of library material.
RNA samples should best be shipped on dry ice. Please only ship with courier services (FedEx, UPS, DHL).
For longer transports (e.g. from South America) we also had very good success with RNA samples shipped dry at room temperature (after LiCL/ethanol precipitation and ethanol washes; see protocol below).
Please mark the position of the pellet on the tubes in this case!
The protocol below is modified from here: http://www.paralog.com/wiki/?EthanolPrecipitation
- Add 1/10th volume of 8M LiCl to your RNA sample and mix.
- Add 3 volumes of room temperature 100% ethanol (based on the aqueous volume before the addition of LiCl) and mix thoroughly.
- Incubate at -20C for 20 minutes or ON.
- Vortex quickly. Mark the side of the tube that will be positioned towards the outside of the centrifuge rotor (the side the pellet will be located).
- Centrifuge at a minimum of 12000g for 15 minutes in a cooled centrifuge (4C).
- Decant the supernatant.
- Add 500 μl room temperature 75% ethanol and rinse the tube by gentle inversion.
- Respin at 12000g for 2 minutes.
- Decant the 75% ethanol.
- Spin down briefly and pipette aspirate the remaining 75% ethanol.
- Repeat the 75% ethanol wash (the previous five steps)
- Allow the pellet to air dry. The sample is now ready for shipping at room temperature.
The samples can then be resuspended in an appropriate volume of molecular biology grade water (best for short term storage and library preps), EB buffer, or TE buffer (only long term storage). Please let us know how much RNA sample to expect per tube.
Please see this FAQ for RNA isolation tips: http://dnatech.genomecenter.ucdavis.edu/faqs/which-protocols-or-kits-do-you-recommend-for-rna-isolations-from-human-and-animal-samples/
- The fragment lengths should be consistent and best be between 100 and 300 bp (up to 400 bp for the majority of molecules is acceptable). Consistent fragment lengths can best be achieved on a Covaris style closed tube sonicator. We recommend avoiding probe sonicators.
- Please make sure to run the input controls on a Bioanalyzer or agarose gel beforehand, and email us an image of these.
- Sequence one “input control” per cell line/sample type.
- Analyze at least two biological replicates.
- We highly recommend verifying the enrichment of your regions of interest (e.g. promoter regions) vs. the control samples by qPCR, before submitting the samples for sequencing.
- For highest accuracy data we can now generate sequencing libraries with UMI-bearing sequencing adapters. UMIs (Unique Molecular Identifiers) allow the accurate detection and removal of PCR duplicate reads. This approach is especially recommended for low-input samples. The first nine bases of the forward and reverse reads will contain UMI sequences.
The required read number per sample will vary from target to target. For the study of point source transcription factors the ENCODE project recommends analyzing at least 20 million (uniquely mapping) reads (http://genome.cshlp.org/content/22/9/1813.long#boxed-text-2). Depending on the quality of your preps, perhaps 75% of the reads can be expected to be uniquely mapping. ENCODE tends to err on the high side with their recommendations. Thus, about 20 million read pairs per sample should be acceptable, but this is likely the minimum number.
Zhang et al. 2016 have studied the impact of the sequencing run types on ChIP-seq data analysis. Their data indicate that paired-end sequencing data provide significant advantages of single-end sequencing in ChIP-seq.
CUT&RUN sequencing might be a better alternative:
CUT&RUN sequencing (Skene & Henikoff 2017) is a faster protocol that for almost all applications is a more sensitive alternative requiring much lower cell numbers. CUT&RUN is suitable for studying histone modifications, transcription factors, and co-factors. In addition to lower input requirements CUT & RUN experiments also afford reduced read numbers (4 to 8 million read pairs per sample).
References:
Landt et al. 2012: ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Research 22: 1813-1831
Bailey et al. 2013: Practical Guidelines for the Comprehensive Analysis of ChIP-seq Data. PLOS Computational Biology https://doi.org/10.1371/journal.pcbi.1003326
Skene & Henikoff 2017: An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. https://doi.org/10.7554/eLife.21856
Zhang et al. 2016: Systematic evaluation of the impact of ChIP-seq read designs on genome coverage, peak identification, and allele-specific binding detection. BMC Bioinformatics volume 17, Article number: 96
MACS — Model-based Analysis of ChIP-Seq https://taoliu.github.io/MACS/
https://hbctraining.github.io/Intro-to-ChIPseq/lessons/05_peak_calling_macs.html
The isolation of high-quality DNA and RNA samples from plants can be challenging due to the presence of inhibiting and damaging phytochemicals. Thus, it is not possible to recommend a single protocol that works for all samples. In any case the RNA samples should be DNAse treated, and QC-ed on a Bioanalyzer for sample integrity and via Nanodrop for purity. Please see sample requirements.
For many species and many types of samples the Qiagen RNeasy Plant MiniKits (cat. no. 74903) have been applied successfully. For RNA-seq and Tag-Seq projects this kit has to be used in conjunction with the Qiagen RNase-Free DNase Set (cat. no. 79254) . We recommend to isolate the RNA, then perform the DNAse digestion on the isolated RNA, and then clean up this reaction once more with the RNeasy kit. Alternatively, you could use the Zymo RNA clean & concentrator 5 kit with DNAse (cat. no. R1013) for the DNAse digestion and clean up.
The NEB Monarch Total RNA Miniprep Kit comes with an “RNA Protection Reagent” that can be added during the mechanical disruption of plant samples.
- Always perform at least two spin column washes (with the kit wash buffer) after binding of the lysed sample to the column matrix. Perform the “optional” steps described in the kit manual. Also, add a short “dry spin” of the column after the washes and before the elution buffer addition to avoid carryover of the ethanol wash buffer.
- Avoid glycogen (often used as a co-precipitant).
- To achieve the cleanest RNA isolations only use at most half the sample amount of the maximum recommended by the manufacturer.
Avoiding Batch-Effects:
Both sample storage conditions and details of the RNA-isolation protocols are well-known to introduce technical variations into RNA-seq data. Because of this, it is recommended to:
- Isolate the RNA-samples in one batch.
- If RNA-isolations need to be carried out in several batches, they should be carried out by the same person using the same batch of reagents
- If RNA-isolations need to be carried out in several batches, the samples should be randomized between the RNA isolation batches (worth discussing with a statistician or the Bioinformatics Core).
- The first PCR is carried out with sequence-specific oligos that are tagged on the 5′-ends with universal tags. The sequences are provided below.
Please note that these first-round primers are amino-modified at the 5′-end to prevent the conversion of unbarcoded amplicons into library molecules. Ordering desalted oligos should be sufficient, as long as the sequence-specific part of the oligo is short (e.g. 20 bp). - The second PCR then uses the universal tags to add sample-specific 16 bp barcode indices to both ends of each amplicon.
REVERSE Tag (U2) 5’-/5AmMC6/TGGATCACTTGTGCAAGCATCACATCGTAG-3’
Sample preparation:
If it has been established that a restriction enzyme (e.g. ApeKI) and method are suitable for the species you are working on (please see below), we require the samples for RR-Seq to be submitted in a 96-well plate. One or two wells should remain empty for negative controls. The concentration of the samples should be normalized to 50 ng/ul as assayed by an intercalating dye (fluorometry using a Qubit; Quantus, or plate reader). To ascertain the chemical purity of the samples, the UV absorption ratios should be 1.8 to 2.0 (260/280 nm) and > 2.0 for 260/230 nm . A volume of 20 ul per sample is sufficient.
The DNA samples have to be extracted using a CTAB-free protocol (best a spin column protocol), since very precise DNA sample quantification is critical for the success of the protocol.
The DNA samples have to be RNA-free. Thus, the DNA isolation protocol has to include an RNAse digestion step.
Before shipping us samples please email us gel images of representative samples. RR-Seq Sequencing is carried out with single-end 100 bp or single-end 150 bp reads.
We offer dual enzyme RR-Seq library sequencing with PstI or SbfI on one end combined with MspI for the other end.
Alternatively, we do offer a restriction-enzyme-free RR-seq protocol which is PCR-based.If there are no sequencing data for these enzymes for your species of interest yet, we would need to establish that the enzymes are suitable to avoid targeting sites present in abundant repeat sequences. We will do this by carrying out test-library preparations (see below).
Many types of RNA-seq require RNA samples of high integrity and high chemical purity – please see the sample requirements. If the tissue or cell samples are handled correctly (e.g. flash frozen and stored at -80C) standard spin column RNA extraction kits will yield RNA samples perfectly suitable for RNA-seq. Please note that samples destined for miRNA or small RNA studies need to be isolated with protocols specifically designed to retain the small molecules (please see below). Standard RNA isolation protocols will lead to the loss and sequence-specific selection of small RNA molecules. RNA samples should always be DNA-free. Nanodrop readings are more or less useless to determine RNA sample concentrations – please use fluorometric quantification instead (e.g. Qubit or Quantus instruments). The Nanodrop readings should be used to assess sample purity.
- For most tissues, the standard Qiagen RNeasy kits (cat. no. 74004) are perfectly fine (or similar kits from other vendors). These RNAeasy kits have to be used in conjunction with the Qiagen RNase-Free DNase kit (cat. no. 79254). We recommend to isolate the RNA, then perform the DNAse digestion in solution on the isolated RNA, and then clean up this reaction once more with the RNeasy kit. Alternatively, a kit like the Qiagen RNeasy Plus Micro Kit (cat. no. 74034) with “DNA Eliminator” gDNA removal columns may be used.
- In case the samples contain interfering chemicals or in case of very small sample amounts, it could be worth trying kits from Norgen Biotek. This manufacturer offers a selection of sample-type specific kits and uses a proprietary silicon carbide spin column matrix, which has a higher affinity for RNA compared to the standard silica columns. Thus, the Norgen Biotek kits often provide higher yields. Norgen kits offer two options for DNA removal: “plus” kits come with a dedicated DNA removal column; standard kits OTOH require DNAse treatment with the DNAse digestion add-on (Norgen cat. no. 25710).
- Always perform at least two spin column washes (with the kit wash buffer) after binding of the lysed sample to the column matrix.
- In case your lab uses a Trizol protocol for RNA isolations, we do recommend an additional sample purification with a spin column kit (e.g. Zymo RNA clean & concentrator 5 kit with DNAse – cat. no. R1013) for a DNAse digestion and clean up.
- An additional column cleanup is mandatory for RNA samples isolated from blood PAXgene or Tempus tubes (the tubes are used for blood preservation) after the initial RNA isolation. Often the preservative chemicals tend to contaminate the samples upon isolation. For the cleanup, we recommend the Zymo RNA clean & concentrator 5 kits with DNAse (cat. no. R1013) or similar.
- For miRNA and small RNA studies, protocols specifically designed to isolate also the shorter molecules have to be employed. Appropriate kits are available from multiple suppliers. Some suitable examples are an array of sample-type-specific NorgenBiotek and Qiagen kits as well as the Zymo Quick RNA kit. Please make sure that you apply the protocol variants designed to retain miRNAs for all of these.
- NEVER use heparin as an anticoagulant for blood samples destined for DNA or RNA sequencing.
- Avoid using glycogen (often recommended as co-precipitant).
- The 260/230 nm and 260/280 nm absorption ratio measurements (e.g. from NanoDrop) are should be used to assess sample purity. For RNA the 260/230nm ratio should be >1.5 and the 260/280nm ratio 1.8-2.1; Please see the sample requirements page and the sample cleanup FAQ.
How many cells will be needed to isolate sufficient RNA for conventional RNA-seq?
The typical mammalian cell contains 10 to 30 pg of RNA. Assuming the worst-case scenario (only 10 pg RNA content; 50% loss during isolation), you should be starting the RNA isolation with 20,000 or more cells to reach at least 100 ng total RNA sample, the lowest amount recommended for RNA-seq after poly-A enrichment. Please see the small sample RNA isolation recommendation above.
Avoiding Batch-Effects:
Both sample storage conditions and details of the RNA-isolation protocols are well-known to introduce technical variations into RNA-seq data. Because of this, it is recommended to:
- Isolate the RNA-samples in one batch.
- If RNA-isolations need to be carried out in several batches, they should be carried out by the same person using the same batch of reagents
- If RNA-isolations need to be carried out in several batches, the samples should be randomized between the RNA isolation batches (worth discussing with a statistician or the Bioinformatics Core).
Related Pages:
04 Library Preparation and QC (16)
Ampure XP bead “upper cut” protocol to remove fragments longer than 670 bases:
-
-
- If not mentioned explicitly follow the standard Ampure XP handling instructions from the manufacturer (e.g. equilibrate the beads at to room temperature before use; vortex beads before use, details of the bead washes and elution,…)
- If the sample volume is smaller than 50 ul, add EB buffer up to 50 ul to each sample.
- Add 0.55x the sample volume in Ampure beads (e.g. 27.5 ul beads to a 50 ul sample) to your sample, mix, incubate for 5 minutes at RT.
- Collect the beads on a magnet.
- Transfer the supernatant to a new tube.
- Add another 1x original volume Ampure beads to the supernatant; mix; incubate for 5 minutes
- Collect the beads on a magnet and remove the supernatant
- Carry out the two regular 80% ethanol washes of the beads and elute the samples from the beads according to Agencourt Ampure XP protocol.
- Verify the success of the size selection by running an aliquot on a Bioanalyzer or equivalent instrument.
-
Ampure XP bead “upper & lower cut” protocol to remove fragments longer than 670 bases and shorter than 400 bases:
This protocol is identical to the one above but adds a smaller volume of the beads at step 6 for the final enrichment onto the beads. The reduced bead buffer concentration at this step leads to a removal of longer fragments compared to the protocol above.
Please note: It is recommended to verify this protocol first with your batch of Ampure XP beads or similar beads from other manufacturers. Bead-based size selection cannot carry out precise “cuts”; thus, you will also lose some of the library in the size ranges that you intend to keep.
- If not mentioned explicitly follow the standard Ampure XP handling instructions from the manufacturer (e.g. equilibrate the beads at to room temperature before use; vortex beads before use, details of the bead washes and elution,…)
- If the sample volume is smaller than 50 ul, add EB buffer up to 50 ul to each sample.
- Add 0.55x the sample volume in Ampure beads (e.g. 27.5 ul beads to a 50 ul sample) to your sample, mix, incubate for 5 minutes at RT.
- Collect the beads on a magnet.
- Transfer the supernatant to a new tube.
- Add another 0.25x of the original volume Ampure beads (e.g. 12.5 ul beads for a sample of a 50 ul starting volume sample) to the supernatant; mix; incubate for 5 minutes
- Collect the beads on a magnet and remove the supernatant
- Carry out the two regular 80% ethanol washes of the beads and elute the samples from the beads according to Agencourt Ampure XP protocol.
- Verify the success of the size selection by running an aliquot on a Bioanalyzer or equivalent instrument.
Beckmann/Agencourt also sells beads that are dedicated to size selections named SPRIselect — however, very likely these are actually identical to the AMpure XP beads. The SPRIselect manual provides a lot of additional information and protocols that can be applied to AMpure XP and other beads. Please see here: Beckman SPRIselect Ampure beads
BTW, our favorite magnetic separator for 96-well plates is this one from EdgeBio.
RNA-seq experiments should best be carried out with samples of consistent RNA integrity and input amounts. However, some RNA-seq samples can be so limited and irreplaceable that experiments have to be carried out with less than the recommended input amounts. Similar complications can occur if some of the samples are significantly more degraded than others. Such situations require weighing the pros and cons when choosing the input amounts from the more abundant samples.
Points to consider are:
- The ideal approach for an RNA-seq project would be to treat each sample exactly the same, to minimize technically induced variation in the resulting data. This would include starting each library prep with the same amount of total RNA input and applying the same number of PCR cycles to each of the libraries. However, more degraded samples usually require increased input amounts.
- In general, sequencing library preparations do not fail at a specific input amount threshold. Lower amounts can usually be compensated for by increasing the number of PCR cycles during the preparation. Thus, inputs lower than the kit manufacturers’ recommendations can be used in some cases. Any reduced input amounts (and/or higher sample degradation) will, however, lead to reduced library complexities, and thus noisier gene expression data. The best data are usually generated when working with input amounts in the upper half of the manufacturers’ input recommendations.
For sample sets with varying RNA sample amounts and qualities, we suggest verifying first if outlier samples with significantly lower sample amounts or lower quality can be dropped from the experiment. If this is not the case we suggest two options. We will ask you to pick one of these or to provide detailed instructions for another approach:
Strategy #1: Normalize all RNA input amounts to the lowest mass sample that has to be included in the study. Please note that this will more severely impact the quality for the originally high RNA quality & high RNA amount samples.
Strategy #2: Normalize the RNA input amounts to a range from the lowest input sample to three times that of the lowest input sample. With this approach, all libraries will still undergo the same number of PCR cycles, which preserves more of the sample quality of the more abundant and higher quality samples. (An example case would be that the lowest available amount for one of the samples is 10 ng. We would then dilute only high-amount samples to an input of at most 30 ng.)
For most projects, we tend to recommend Strategy #2, especially if the ratio of the low-input outlier samples is low.
Is PCR-free library preparation still advantageous?
In general, the original concerns about library PCR amplification (presented in papers from 2008) are no longer very relevant. This is due to the use of modern polymerases that are designed for complex samples like Kapa HiFi, NEB Q5, or QIAseq HiFi polymerase. The previous “standard”, the high-fidelity Phusion enzyme had tremendous disadvantages for complex samples (Quail et al. 2012 Optimal enzymes for amplifying sequencing libraries. Nature Methods volume 9, pages10–11(2012) https://www.nature.com/articles/nmeth.1814 ).
PCR-free libraries also have disadvantages, since they require significantly higher library QC efforts. Thus, we are charging a PCR-free Add-On fee for the preparation of PCR-free libraries.
What are your recommendations?
A great alternative to preparing the libraries completely PCR-free is the use of a single PCR cycle instead. This combines the advantages of both: It creates fully double-stranded library molecules that do not cause any problems in the library QC. In addition there will be no or only an extremely low PCR-bias introduced. Our recommendation is to submit the same amount of DNA sample as for PCR-free library preps (e.g. 1 ug) and then request the single PCR cycle library amplification.
Quality and quantity of DNA and RNA is critical for high quality sequencing output. Please make sure your DNA is not degraded and is free of RNA contamination. RNA samples should always be assessed on the bioanalyzer for the absence of gDNA contamination (can be removed with DNaseI treatment followed by a column clean-up; e.g. Zymo “RNA Clean and Concentrator”) and degradation. Preferentially determine the concentrations of your DNA and RNA samples using fluorometry (e.g. with a Qubit or plate reader). The sample purity should be assessed by spectrophotometry (e.g. Nanodrop). Please see this page for a comprehensive table of sample requirements for sample QC, library preps, or your self-made libraries. Please see the Library Prep Page for details on the library prep processes. For submission information, including submission forms and shipping details, please visit the Sample Submission & Scheduling page. If you are submitting DNA for PacBio libraries, please follow the PacBio Guidelines for Shipping and Handling.
The Real-time PCR core can carry out DNA as well as RNA extractions for you.
When designing RNA-seq or ChIP-seq experiments, it is very important to avoid technical replicates and pseudo-biological replicates as they will lead to spurious results (e.g. spurious differential gene expression data; DGE data in case of RNA-seq).
Creating pseudo-biological replicates occurs frequently, especially for in vitro studies. Doing so can often lead to hundreds of false positive differentially expressed genes. For example, treating three cell-culture flasks of the same passage of a cell line as biological replicates would create such a dilemma. Please see the excellent discussion of this topic by Christoph Emmerich here: https://paasp.net/accurate-design-of-in-vitro-experiments-why-does-it-matter/ .
This video by Josh Starmer explains why technical replicates are not helpful in principle in RNA-seq: https://www.youtube.com/watch?v=gKnfP2_Xdpo .
Ampure XP/SPRI bead “upper cut” protocol to remove double-stranded DNA fragments over 670 bases:
- Bead-based size selection cannot carry out precise “cuts”; Thus, you will also lose some of the library molecules in the size ranges that you intend to keep. This selection protocol will also reduce adapter dimers and other molecules shorter than 160 bp.
- It is recommended to verify this protocol first with your batch of beads.
- Multiple other manufacturers offer copies of the Ampure XP product (e.g. SPRI beads, Kapapure, …). These can work just as efficiently. Please test them beforehand.
- The cutoff fragment length can be modified by changing the ratios of SPRI-beads to sample volume.
-
-
- If not mentioned explicitly follow the standard Ampure XP handling extractions from the manufacturer (e.g. equilibrate the beads to room temperature before use; vortex beads before use, details of the bead washes and elution,…)
- If the sample volume is smaller than 50 microliters, add molecular biology grade water up to 50 microliters.
- Add 0.55x the sample volume in Ampure XP beads to your sample, mix, incubate for 5 minutes at RT.
- Collect the beads on a magnet.
- Transfer the supernatant to a new tube.
- Add another 1x original volume Ampure beads to the supernatant; mix; incubate for 5 minutes
- Collect the beads on a magnet and remove the supernatant
- Carry out the regular 80% ethanol washes of the beads and elute the samples from the beads according to Agencourt Ampure XP protocol.
- Verify the success of the size selection by running an aliquot on a Bioanalyzer or equivalent instrument.
-
In case the library preparation did not generate sufficient library material required to load a sequencer, Illumina libraries can be amplified with a universal PCR protocol.
While the amplification can rescue experiments, it is worth considering on a per-project basis if perhaps the library preparation should be repeated instead. For quantitative experiments, it is generally recommended to treat all libraries the same throughout the pipeline. Insufficient library yields in the initial library preparation could be signs of sample contamination, processing errors, etc. with potential side effects that cannot be remedied by amplification.
Illumina library amplification PCR protocol:
The library amplification uses standard Illumina P5 (5′-AATGATACGGCGACCACCGAGATCT-3′) and P7 (5′-CAAGCAGAAGACGGCATACGAGAT-3′) PCR primers which can be ordered as desalted DNA oligos.
Create a 10x concentrated primer mix at 10 μM each of these primers in EB buffer.
Add up to 20μl library, add water up to a volume of 20μl, add 5 μl 10x primer mix. Then add 25μl Kapa HiFi 2x Hotstart PCR master mix and pipette up and down several times.
Use the following cycling parameters:
Initial denaturation: 98°C 45 sec,
X PCR amplification cycles consisting of: denaturation 98°C 15 sec, annealing 60°C 30 sec, extension 72°C 30 sec
Final extension 1 min.
Assuming one wants to generate at least 100 ng of sequencing library, we recommend performing four cycles of PCR when starting from 10 ng library.
After the PCR perform a standard Ampure XP/SPRI bead cleanup (e.g. with beads at 1.2x the sample volume, Ampure beads or equivalent) and elute in 30μl EB buffer.
3’Tag-Seq is a protocol to generate low-cost and low-noise gene expression profiling data. The protocol is also known as TagSeq, 3’Tag RNA-Seq, Digital RNA-seq, Quant-Seq (please note that most of these names have also been used for a variety of other protocols previously). In contrast to traditional RNA-Seq, which generates sequencing libraries from the whole transcripts, 3-Tag-Seq only generates a single initial library molecule per transcript, complementary to 3′-end sequences. For example for human samples, the restriction to a small part of the transcripts reduces the number of sequencing reads required by at least five times. In contrast to earlier “digital RNA-seq” protocols that were based on restriction digestions of cDNAs, the current protocol combines reverse transcription priming from the poly-A tail with random priming and adapter placement for the second-strand synthesis. In most cases up to 48 samples can be sequenced per HiSeq 4000 lane.
More than 90% of the RNA-seq studies carried out in our labs are analyzed exclusively for differential gene expression (DGE). The conventional full transcript RNA-seq protocols generate more data than needed for this specific purpose, but they also allow for splicing analyses. The complexity of the standard RNA-seq data is not an advantage if the aim of the project is only DGE analysis – 3’Tag-Seq might actually be the superior tool for this application (DGE). In our experience the 3’Tag-Seq data have so far shown exceptionally low noise as well as insensitivity to RNA sample quality variations.
This example MDS plot shows an analysis of 3’Tag-Seq data of macrophage cells exposed to three types of bacterial infections and mock-infections at two time points. The analysis distinguishes the responses to the individual bacterial species and the duration of the infections. Even the reactions to the mock-infections are clustered by time points.

We are currently offering 3’Tag-Seq as a low cost custom sequencing service but are planning to offer 3’Tag-Seq services soon at simple per-sample recharge rates — including both library preps and sequencing. In the long run the services can also include a basic differential-gene-expression analysis.
Advantages of 3’Tag-Seq:
- low noise gene expression profiling
- less sensitive to RNA sample quality/integrity variations (compared to poly-A enrichment protocols)
- >99% strand-specific; same direction as mRNA transcripts
- requires significantly lower numbers of sequencing reads
- single read sequencing is sufficient
- simpler library prep protocol
- costs about half or less compared to standard RNA-seq
- costs lower than, or comparable to, microarray analysis
- much higher dynamic range compared to microarrays
- we routinely sequence 48 libraries per HiSeq lane; for soBarclays
- for very low input or high depth sequencing of 3’Tag-Seq libraries UMI‘s (unique modular identifiers) can be incorporated
- Batch-Tag-Seq packages: simple pricing scheme and simplified planning of experiments
Disdavantages of 3’Tag-Seq:
- data analysis requires a reference genome with good annotation (including UTRs)
- only applicable to eukaryotic samples
- data do not contain any transcript-splicing information
- protocol is (a bit) more sensitive to chemical contaminants (spin column cleaned RNA samples are recommended)
For high-throughput 3’Tag-Seq library generation we require pure total RNA samples at a concentration of 100 ng/ul (best submit 10 ul at 100 ng/ul). For custom 3’-Tag-Seq library preps the input amounts can be a low as 10 ng total. The RNA samples for this protocol need to be isolated or cleaned-up by spin-column protocols. Please also see the sample requirements page.
3′-Tag-Seq libraries are sequenced by single-end sequencing on the HiSeq 4000 or the NextSeq.
Please note that 3’Tag-Seq libraries generate lower read numbers on the HiSeq 4000 (about 320 million reads per lane) compared to standard RNA-seq libraries. Since the DGE analysis of tag-Seq data requires much lower read numbers this is usually not a problem.
The libraries will be sequenced on Illumina HiSeq 4000 or NextSeq 500 sequencers with single-end 80 or 90 bp reads (SE80 or SE90). Please note that for some analysis pipelines it is recommended to trim off the first 12 bases from the reads. We will provide the full length data. Trimming is not necessary if you are using a local aligner (like STAR or BBmap). The sequences can be trimmed easily, for example with the “reformat” command from BBTools. In case UMIs are incorporated, the first 6 bases of the forward read represent the UMI, followed by a common linker with the sequence “TATA”, followed by the 12bp random priming sequence. It is recommended to transfer the UMI sequence information to the read header and trim the first 22 bases from each read with UMI-TOOLS or custom scripts. The same software can be used to remove PCR-duplicates after the alignments.
Please also the 3’Tag-Seq data analysis recommendations and this note on working with degraded RNA samples.
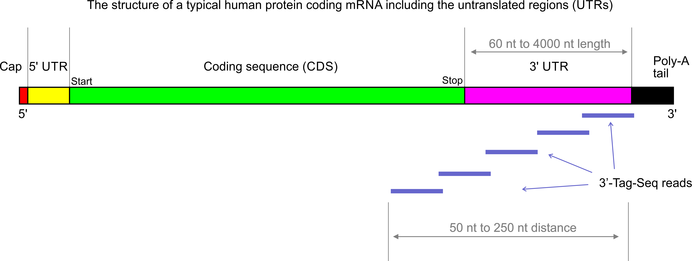
Comprehensive Batch-Tag-Seq info for download with UC pricing.
Primer and adapter-dimer contamination in sequencing libraries can lead to serious problems like barcode switching (also called barcode hopping). Thus, these short molecules should be removed from the libraries as soon as traces of them become visible on the Bioanalyzer or equivalent. Please note that the Bioanalyzer uses double-strand-specific fluorescent dyes which have a very low affinity to single-stranded primers. Thus, the Bioanalyzer assay severely underestimates the true concentration of free primer molecules.
- Low concentrations of free primers and adapter-dimers can be removed with a bead cleanup e.g. adding 1x the original volume in Ampure XP beads (or equivalent).
- The more stringent option for primer removal is an Exonuclease VII single-strand digest (e.g. with this ExoVII) at 37C for 20 minutes using 1 ul enzyme and the accompanying buffer; followed by a bead cleanup with 1.6 x the original volume in Ampure XP beads. This will not remove primer-dimers.
PCR amplified sequencing libraries frequently display library molecules seemingly about twice the excepted size or even bigger. In most cases, this phenomenon is caused by over-amplification of the libraries. These PCR artifacts do occur in cases the PCR reactions run out of essential reagents – in most cases the PCR primers will be exhausted. If primers are no longer available the PCR products will anneal to each other (the sequencing adapter sequence will be the by far most common sequences available). The resulting annealing products are often called “PCR-bubbles” and are partly double-stranded and partly single-stranded; thus they migrate considerably slower on agarose gels as well as on Bioanalyzer assays. Please see below.
Since these artifacts are merely annealing products, the resulting libraries are perfectly sequence-able. However, the quantification of such libraries by fluorometry will not be precise since the dyes used for these measurements are specific for double-stranded DNA molecules and PCR bubbles contain considerable amounts of single-stranded DNA that will not be measured. The PCR bubbles can be removed by amplifying the library one more time with a single cycle of PCR (a so-called “Reconditioning PCR“). For this PCR you could use standard Illumina P5 and P7 primers (please see below). The complementary sequences should be located at the very ends of all Illumina sequencing library molecules. In most cases PCR bubble artifacts can not be removed by SPRI bead size selections or Blue Pippin size selections; if necessary, a “Reconditioning PCR” is the best option.
However, to avoid unnecessary complexity loss of the library and introduction of polymerase errors, it would be best to optimize the library preparation protocol for a lower number of PCR cycles beforehand.
Reconditioning PCR protocol:
The reconditioning PCR uses standard Illumina P5 (5′-AATGATACGGCGACCACCGAGATCT-3′) and P7 (5′-CAAGCAGAAGACGGCATACGAGAT-3′) PCR primers which can be ordered as desalted DNA oligos.
Create a 10x concentrated primer mix at 20 μM each of these primers in EB buffer.
Add 1 to 4 μl previous PCR product, 2 μl 10x primer mix, 3 to 7 μl water for a total volume of 10 μl, then add 10 μl Kapa HiFi 2x Hotstart PCR master mix and pipette up and down several times.
Use the following cycling parameters: Initial denaturation 98°C 45 sec, denaturation 98°C 15 sec, annealing 60°C 30 sec, extension 72°C 30 sec, final extension 1 min.
Perform a standard Ampure XP/SPRI bead cleanup (e.g. with beads at 1.2x the sample volume) before analyzing the product by microcapillary electrophoresis.
Another graphic illustrating PCR bubbles (source Illumina Inc.). Please also see: https://support.illumina.com/bulletins/2019/10/bubble-products-in-sequencing-libraries–causes–identification-.html
Illumina sequencers using the patterned flowcell technology (NovaSeq X, iSeq, NextSeq 2000, HiSeq 4000 &X Ten) can show an increased rate of barcode switching events. These artifacts are enabled by the exclusion amplification chemistry used on these sequencers; IIlumina calls the artifacts “index hopping”. Please see the index hopping information from Illumina here and in this video from Illumina. We have also have posted additional information here.
Please note that the AVITI sequencers, the MiSeq sequencer and NextSeq 500 chemistries are not sensitive to index hopping. For these sequencers single-indices and combinatorial indexing are fine.
To best avoid barcode switching read mis-assignments on new Illumina sequencers two measures are recommended:
- The use of uniquely dual-indexed (UDI) adapters. Illumina has collected information on the design of such adapters here. This adapter design employs unique barcodes for both index 1 and index 2 for each of the pooled libraries. Thus, 96 samples will use 96 different index 1 adapter sequences and 96 different index 2 adapter sequences.
- The efficient removal of any free primers and adapter-dimers from the libraries
The use of UDI adapters is highly recommended especially for the NovaSeq. Hereby, matching i5 and i7 indices per library must be avoided. UDI adapters can also be used on the MiSeq and the NextSeq, but they do not offer any significant advantages on these sequencers that employ the bridge-amplification chemistry. Further, any traces of free primers, primer-dimers, and adapter dimers should be removed from the sequencing libraries or the pools.
Commercial sources of UDI adapters: TruSeq-style uniquely indexed adapters are available from both Illumina and BiooScientific. Qiagen, NEB, and NuGEN are also supporting their library prep kits with optional UDI adapters. Nextera style indices need to be custom ordered from oligo vendors.
The DNA Technologies Core has 96-plex UDI adapter sets in stock that can be added to sequencing libraries by PCR. These barcode sets are available for both Nextera and TruSeq adapter designs. Please note that for TruSeq style libraries one will need to ligate a shortened and index-less stub-adapter instead of a standard Illumina adapter. The indices are then added after the cleanup of the ligation reaction by PCR.
Sequencing libraries prepared by the DNA Technologies Core:
The DNA Technologies Core uses UDI adapters for all library prep protocols that are compatible with dual indexing (e.g. DNA-Seq, RNA-seq, 3′-Tag-Seg, WGBS-Seq, …). The Core also makes sure to remove any traces of free primers, primer-dimers, and adapter-dimers.
- The fragment lengths should be consistent and best be between 100 and 300 bp (up to 400 bp for the majority of molecules is acceptable). Consistent fragment lengths can best be achieved on a Covaris style closed tube sonicator. We recommend avoiding probe sonicators.
- Please make sure to run the input controls on a Bioanalyzer or agarose gel beforehand, and email us an image of these.
- Sequence one “input control” per cell line/sample type.
- Analyze at least two biological replicates.
- We highly recommend verifying the enrichment of your regions of interest (e.g. promoter regions) vs. the control samples by qPCR, before submitting the samples for sequencing.
- For highest accuracy data we can now generate sequencing libraries with UMI-bearing sequencing adapters. UMIs (Unique Molecular Identifiers) allow the accurate detection and removal of PCR duplicate reads. This approach is especially recommended for low-input samples. The first nine bases of the forward and reverse reads will contain UMI sequences.
The required read number per sample will vary from target to target. For the study of point source transcription factors the ENCODE project recommends analyzing at least 20 million (uniquely mapping) reads (http://genome.cshlp.org/content/22/9/1813.long#boxed-text-2). Depending on the quality of your preps, perhaps 75% of the reads can be expected to be uniquely mapping. ENCODE tends to err on the high side with their recommendations. Thus, about 20 million read pairs per sample should be acceptable, but this is likely the minimum number.
Zhang et al. 2016 have studied the impact of the sequencing run types on ChIP-seq data analysis. Their data indicate that paired-end sequencing data provide significant advantages of single-end sequencing in ChIP-seq.
CUT&RUN sequencing might be a better alternative:
CUT&RUN sequencing (Skene & Henikoff 2017) is a faster protocol that for almost all applications is a more sensitive alternative requiring much lower cell numbers. CUT&RUN is suitable for studying histone modifications, transcription factors, and co-factors. In addition to lower input requirements CUT & RUN experiments also afford reduced read numbers (4 to 8 million read pairs per sample).
References:
Landt et al. 2012: ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Research 22: 1813-1831
Bailey et al. 2013: Practical Guidelines for the Comprehensive Analysis of ChIP-seq Data. PLOS Computational Biology https://doi.org/10.1371/journal.pcbi.1003326
Skene & Henikoff 2017: An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. https://doi.org/10.7554/eLife.21856
Zhang et al. 2016: Systematic evaluation of the impact of ChIP-seq read designs on genome coverage, peak identification, and allele-specific binding detection. BMC Bioinformatics volume 17, Article number: 96
MACS — Model-based Analysis of ChIP-Seq https://taoliu.github.io/MACS/
https://hbctraining.github.io/Intro-to-ChIPseq/lessons/05_peak_calling_macs.html
We have currently 96 indices are available and can pool 96 RNA-seq or genomic sequencing libraries. Bioo Scientific offers NEXTflex barocde sets allowing the pooling of up to 384 libraries. If you are planning to use homebrew versions of indices please consult with us first, as reduced complexity from incorrectly designed indices may cause failures when sequencing your sample.
If you have access to fluorometric DNA quantification and a Bioanalyzer (or equivalent), library pooling is not difficult. We offer the pooling of sequencing libraries for a small fee. For sequencing libraries generated by the Core, pooling is included in the library preparation service.
Prerequisites for the pooling of customer libraries are:
- all libraries were generated using the same protocol and are PCR amplified
- the library fragment sizes have to be similar for all libraries * (and within Illumina specs) as demonstrated by Bioanalyzer traces (or gel images if correct balancing is not that critical)
- have uniquely indexed adapters
- all libraries have DNA concentrations in the same range
- PCR-amplified libraries can be quantified based on fluorometric measurements (e.g. Qubit), but PCR-free libraries are best quantified by qPCR.
Library pooling requires precise pipetting of very small volumes. Even with the best libraries, there will be some imbalances. However, and we can’t work magic with variably sized samples.
* The clustering efficiency of Illumina sequencing libraries varies with the fragment lengths. Shorter molecules are more mobile and will always cluster preferentially compared to longer molecules (smaller molecules will “win the race” to the flowcell surface oligos). Thus, accurate pooling is impossible when combining libraries of varying lengths and we can’t vouch for the results. In some cases, it is advisable to size-select the libraries stringently before quantification and pooling.
For sequencing libraries generated by the Core, pooling is included in the library preparation service.
We suggest the following procedure when pooling libraries yourself:
- verify that Bioanalyzer traces of your libraries show the same fragment size distribution
- quantify each library by fluorometry (Qubit or plate reader)
- if necessary dilute some of the highly concentrated libraries (to bring them in line with the others)
- re-quantify the newly diluted libraries (Qubit)
- Under the precondition, that all libraries show very similar fragment size distributions there are in principle two options:
#1 Dilute all libraries to the same concentration, re-quantify the libraries, then pool the same volumes. This option is more laborious than #2, but allows more precise re-pooling if needed (since all libraries have similar concentrations at least).
#2 Pool the same a amounts of each library based on the first round of quantifications. This means pooling varying volumes. As long as the libraries are consistent in length this could mean pooling the same amounts in ng. If the length is a bit variable one should pool the same number of femtomoles for each library; the femtomole amount is calculated by multiplying the concentration (in nM) by volume (in ul). The molarity calculation will consider the fragment lengths and compensate for varying length. But see tip 3) below !!! - Quantify the resulting pool by Qubit to verify that it has the expected concentration (we will quantify once more by qPCR before sequencing)
Please note that the combined library concentration of the pool should be 5 nM or higher (Illumina sequencers) or 16 nM or higher (AVITI); this means that the concentration of individual libraries in the pool can and will be considerably lower.
You can get trained and use a Qubit in our lab: http://dnatech.genomecenter.ucdavis.edu/qubit-fluorometer/
Three more tips for library pooling:
1) Best pipette volume ranges that allow the pipetting to be reproducible and accurate while still saving some library. For example aiming for a volume range from 3 ul to 10 ul.
Likely you will pool much more library than is needed in the end for sequencing.
2) In case you chose option #2 above and your library concentrations vary more than +- 60%, it is often helpful to work with two sub-pools: One for the higher concentration samples and one pool for the lower concentration samples.
The example numbers here are arbitrary. Under the precondition, that all libraries show very similar fragment size distribution, one would for example pipette 100 ng of each high-concentration library into one tube (Pool A) and 20 ng of each low-concentration library into another tube (Pool B). Vortex mix and spin down each of the tubes twice.
For the final combined pool one could then use the complete volume of Pool B and add one fifth of the total volume of Pool A. Vortex and spin down the combined pool twice. The final combined pool thus should contain 20 ng for each of the libraries aiming for balanced sequencing data. (Please note that the library amounts and pooling ratios mentioned here are only examples and that you have to choose appropriate ratios based on the library concentrations of your experiment).
3) If the fragment length distribution of the libraries is variable, one should try to adjust for these length differences (convert the library concentrations into molarities for this purpose). Further consider that shorter library molecules will be faster and cluster preferentially. To generate similar read numbers, one has to pool more library molecules for longer library molecules. Obviously more guessing and a bit of gambling will be involved in this case and pooling will be less accurate.
Strand-Specific RNA-Seq Libraries
RNA-Seq (conventional) after Poly-A enrichment or ribodepletion:
By default we generate strand-specific RNA-seq libraries. Strand-specific (also known as stranded or directional) RNA-seq libraries substantially enhance the value of an RNA-seq experiment. They add information on the originating strand and thus can precisely delineate the boundaries of transcripts in regions with genes on opposite strands.
There are several ways to accomplish strand-specificity. We incorporate dUTP during the second-strand synthesis of the cDNA. The dUTP containing strand will not be amplified by the proofreading polymerase used for library amplification, thus preserving the strand information for RNA-seq.
For single-end sequencing, the resulting data will represent the “anti-sense strand”. When using paired-end sequencing, the forward read of the resulting sequencing data represents the “anti-sense strand” and the reverse read the “sense strand” of the genes (for Trinity transcriptome assemblies the “–RF” orientation flag should be used). Illumina paired-end reads are always inward oriented (with the exception of “jumping” or “mate-pair” libraries).
Tag-Seq:
Tag-seq data are strand-specific and have a “sense-strand” orientation.
small-RNA-Seq/miRNA-seq:
Small RNA-seq data are strand-specific. The forward read of the sequencing data (read 1) is oriented as the reverse complement of the original RNA molecule. For libraries generated with the Revvity (PerkinElmer) Nextflex small RNA kits, the RNA sequence will be both preceded and followed by four bases with a random sequence.
Illumina sequencing libraries are usually generated with Y-adapters. These are partly single-stranded and partly double stranded.
A PCR-free library will thus still contain partly single-stranded regions. These single-stranded regions can lead to several types of Bioanalyzer artifacts. Most commonly the libraries will appear about 70 to 100 nucleotides longer than expected. However, we have also encountered PCR-free libraries that ran as shorter molecules as well as dramatically longer molecules. We have (very rarely) encountered another significant problem: considerable amounts adapter-dimers were not visible on the Bioanalyzer traces of PCR-free libraries.
To accurately QC PCR-free Illumina libraries we recommend the following approach:
– Take a 1 ul aliquot of your library and run a short PCR (e.g. 6 cycles) with this aliquot.
– Clean up the PCR reaction with a spin column ( e.g. Qiagen Qiaquick, Zymo DNA -clean, …); do NOT use Ampure beads.
– Run the cleaned up PCR product on the Bioanalyzer again as well as the original PCR-free library.
The Bioanalyzer trace of the PCR product will represent the true molecule sizes and the true adapter-dimer content the closest.
Is PCR-free library preparation still advantageous?
If we generate PCR-free libraries in our lab, the described additional QC steps for PCR-free libraries will necessitate significant additional costs for the library preparation. Please see the prices for the PCR-free Add-on.
A great alternative to preparing the libraries completely PCR-free is the use of a single PCR cycle instead. This combines the advantages of both: fully double-stranded library molecules can be used for the library QC and there will be no or only an extremely low PCR-bias introduced. Our recommendation is to submit the same amount of DNA sample as for PCR-free library preps (1 ug or more) and then only apply the single cycle of amplification.
In general, the original grave concerns about library PCR amplification (presented in papers from 2011) are no longer very relevant. This is due to the use of modern polymerases that are designed for complex samples like Kapa HiFi, NEB Q5, or QIAseq HiFi polymerase. The previous “standard”, the high-fidelity Phusion enzyme had tremendous disadvantages for complex samples (Quail et al. 2012 Optimal enzymes for amplifying sequencing libraries. Nature Methods volume 9, pages10–11(2012) https://www.nature.com/articles/nmeth.1814 ).
05 Sequencing (18)
Depending on sequencer and in case of the HiSeq 4000 even depending on run type (single-end or paired-end) Illumina uses different approaches to sequence the indices. Please find detailed information here: indexed-sequencing-overview-guide-15057455-04-Illumina-pages1to8
The correct orientation of the barcode sequence fuehrer depends on the way the barcodes are added to the library. The gist of it is:
For barcodes added to Illumina libraries via a PCR step (e.g. Nextera; or also onto TruSeq stub-adapters):
- index 1 (i7) is always read as reverse complement of the sequence in TruSeq or Nexterastyle PCR oligos
- index 2 (i5) is read in direction of TruSeq or Nextera PCR oligos for Miseq, HS4000 SE (single-end), and NovaSeq
- index 2 (i5) is read as reverse complement of barcoded PCR oligos for NextSeq, iSeq, and HS4000 PE runs
For barcoded adapters added via ligation (e.g. standard Truseq style Y-adapters):
- index 1 (i7) is always read in direction(5’to 3′) of the sequence in TruSeq style oligo
- index 2 (i5) is read in direction of TruSeq or Nextera PCR oligos for Miseq, HS4000 SE (single-end), and NovaSeq
- index 2 (i5) is read as reverse complement of barcoded PCR oligos for NextSeq, iSeq, and HS4000 PE runs
Unfortunately, we cannot demultiplex inline barcodes.
Depending on sequencer model, the second index (called i5 index) will be read in different orientations. The correct adapter sequence information for the sample sheets is usually provided for both options by kit manufacturers. The two sequences are reverse complements of each other.
- The Forward Strand Workflow (previously known as Workflow A) sequences are required for MiSeq and AVITI.
- The Reverse Strand Workflow (previously Workflow B) sequences are required for NovaSeqs and NextSeq.
BTW, the AVITI demultiplexing software is smarter and can figure out the correct orientation on its own. Either orientation will be fine for AVITI submissions.
Here is the complete information on this topic from Illumina: https://support-docs.illumina.com/SHARE/IndexedSeq/indexed-sequencing.pdf
Certainly. Please provide a Bioanalyzer profile (we can also generate these), and barcode sequence information in the sample submission form. We will check the quantity of your libraries using real-time PCR (included in the sequencing price). We suggest to submit your library in at least 15 ul volume at a minimum concentration of 5 nM. Please see the Sample Requirements page for details. Depending on the sequencing platform, we can work with less library (down to 1 nM), but the quantification becomes less reproducible, the library becomes less stable, and relatively larger amounts of the library get lost sticking to the tube. The best buffer to store and submit libraries is 10 mM Tris/0.01% Tween-20 ph=8.0 or 8.4, but EB buffer is also acceptable. Please use 1.5 ml low-retention tubes ( e.g. Eppendorf DNA LoBind). If you do not provide a Bioanalyzer profile of your library, we will carry out the QC for a fee.
Please note that for the HiSeq3000/HiSeq4000 the libraries should have fragment lengths not longer than 550 bases and few molecules longer than 670 bp.
UMI is an acronym for Unique Molecular Identifier. UMIs are complex indices added to sequencing libraries before any PCR amplification steps, enabling the accurate bioinformatic identification of PCR duplicates.
UMIs are also known as “Molecular Barcodes” or “Random Barcodes”. The idea seems to have been first implemented in an iCLIP protocol (König et al. 2010).
UMIs are valuable tools for both quantitative sequencing applications (e.g. RNA-Seq, ChIP-Seq) and also for genomic variant detection, especially the detection of rare mutations. UMI sequence information in conjunction with alignment coordinates enables grouping of sequencing data into read families representing individual sample DNA or RNA fragments. Please see the graphic below.
The problems UMIs are addressing:
– Quantitative analysis: Many sequencing library preparation protocols enable high-throughput sequencing (HTS) from low amounts of starting material. Their preparation requires PCR amplification of the libraries. While the PCR polymerases and reagents have been improved greatly in recent years enabling a mostly unbiased amplification of sequencing libraries, some biases still remain against sequences with extreme GC contents and against long fragments. When starting from ultra-low input samples, stochastic effects in the first rounds of the PCR add to the problems. These issues can potentially cause erroneous quantitation data. Removal of PCR duplicates using alignment coordinate information is especially inefficient such for low input situations but also for deep sequencing data. In the latter case alignment coordinate-based de-duplification will remove large numbers of biological duplicate reads from the data, especially for the most abundant transcripts.
UMIs alleviate the PCR duplicate problem by adding unique molecular tags to the sequencing library molecules before amplification.
Please also see our FAQ: “Should I remove PCR duplicates from my RNA-seq data?” for more information.
– Rare variant analysis: Illumina sequencing provides data with low error rates (~0.1 to 0.5%) for most applications. These low error rates nevertheless interfere with the confident identification of low abundance variants. UMI-less data can’t distinguish between these and sequencing errors. UMIs in combination with deep sequencing yielding multiple reads for each of the sample DNA fragments solved this problem. The approach was first described as Duplex Sequencing. Hereby, single-strand consensus sequences (SSCSs) and Duplex consensus sequences (DCSs) assembly of the read families increase the accuracy of the sequencing data significantly. Please note that the DNA sample starting amounts and the library yields have to be controlled for this approach to be efficient. Applications include sequencing of heterogeneous tumor samples, cfDNA sequencing including ctDNA sequencing, deep exome sequencing.
The usage of UMIs is recommended primarily for three scenarios: very low input samples, very deep sequencing of RNA-seq libraries (> 80 million reads per sample), and the detection of ultra-low frequency mutations in DNA sequencing. For many other types of projects, UMIs will yield minor increases in the accuracy of the data. In addition, UMI analysis is an excellent QC tool of library complexity.
Incorporating UMIs into sequencing libraries:
– Our 3′-Tag-RNA-Seq protocols employ UMIs by default . For Tag-seq the first 6 bases of the forward read represent the UMI. These are followed by a common linker with the sequence “TATA”, followed by the 12 bp random priming sequence. It is recommended to transfer the UMI sequence information to the read header and to trim the first 22 bases from each read with UMI-TOOLS or custom scripts.
– For conventional RNA-seq and DNA sequencing applications you will specifically have to request UMIs on the submission form. The default library preparations will NOT use UMIs. The UMIs will be located in-line with the insert sequences for conventional RNA-seq, genomic DNA-sequencing, or ChIP-seq. The first twelve bases of both forward and reverse reads will represent UMIs and associated linker sequences (7 nt UMI sequence followed by a 5 nt spacer “TGACT”; UMIs of forward and reverse read are independent resulting in a combined UMI length of 14nt). UMIs and spacer are then followed by the biological insert sequences (for paired-end data a total of 22 bp will be dedicated to the UMIs instead of the inserts). The UMI and spacer sequences are usually trimmed off and the information transferred into the read ID header with software utilities like UMI-Tools or FASTP.
The figure below displays the (simplified) principles of the UMI data analysis for quantitative and variant detection studies.
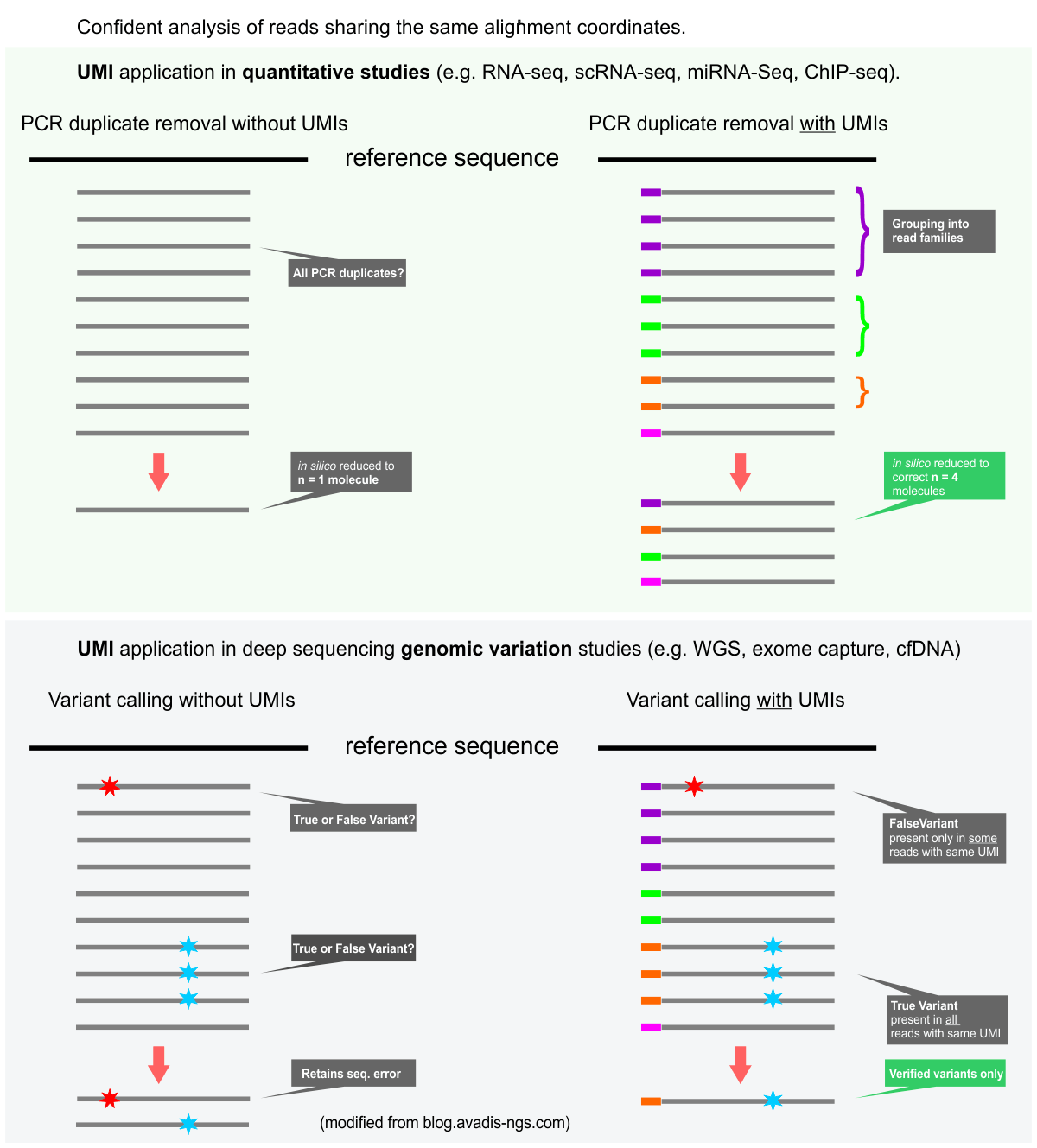
References:
Parekh et al 2016: The impact of amplification on differential expression analyses by RNA-seq. and
Fu et al. 2018: Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers.
Kennedy et al. 2015: Detecting ultralow-frequency mutations by Duplex Sequencing.
König et al. 2010: iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution.
Smith et al. 2017: UMI-tools: Modelling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy.
Software:
UMI-Tools: https://github.com/CGATOxford/UMI-tools
zUMIs: https://github.com/sdparekh/zUMIs
fastp: https://github.com/OpenGene/fastp (transfer of UMIs into read IDs)
Illumina has posted a Beginners Guide on their technology at:
https://www.illumina.com/science/technology/next-generation-sequencing/beginners.html
Please also see our information at: https://dnatech.genomecenter.ucdavis.edu/illumina-high-throughput-sequencing/
The Illumina specifications are based on the Illumina PhiX control library. Better or similar yields can be expected for other high complexity libraries (e.g. genomic, RNA-seq libraries) if they are within the recommended insert size ranges and do not average extreme GC-contents. Yields can vary depending on library type. For libraries that fulfill the criteria above, we do promise that the Hiseq 4000 and NextSeq sequencing data will exceed the Illumina yield specifications.
The table below displays the read numbers as CPF (clusters passing filter). For single-end sequencing CPF is equal to the read numbers. In case of paired-end sequencing the read number is twice the CPF figure.
PCR amplified sequencing libraries frequently display library molecules seemingly about twice the excepted size or even bigger. In most cases, this phenomenon is caused by over-amplification of the libraries. These PCR artifacts do occur in cases the PCR reactions run out of essential reagents – in most cases the PCR primers will be exhausted. If primers are no longer available the PCR products will anneal to each other (the sequencing adapter sequence will be the by far most common sequences available). The resulting annealing products are often called “PCR-bubbles” and are partly double-stranded and partly single-stranded; thus they migrate considerably slower on agarose gels as well as on Bioanalyzer assays. Please see below.
Since these artifacts are merely annealing products, the resulting libraries are perfectly sequence-able. However, the quantification of such libraries by fluorometry will not be precise since the dyes used for these measurements are specific for double-stranded DNA molecules and PCR bubbles contain considerable amounts of single-stranded DNA that will not be measured. The PCR bubbles can be removed by amplifying the library one more time with a single cycle of PCR (a so-called “Reconditioning PCR“). For this PCR you could use standard Illumina P5 and P7 primers (please see below). The complementary sequences should be located at the very ends of all Illumina sequencing library molecules. In most cases PCR bubble artifacts can not be removed by SPRI bead size selections or Blue Pippin size selections; if necessary, a “Reconditioning PCR” is the best option.
However, to avoid unnecessary complexity loss of the library and introduction of polymerase errors, it would be best to optimize the library preparation protocol for a lower number of PCR cycles beforehand.
Reconditioning PCR protocol:
The reconditioning PCR uses standard Illumina P5 (5′-AATGATACGGCGACCACCGAGATCT-3′) and P7 (5′-CAAGCAGAAGACGGCATACGAGAT-3′) PCR primers which can be ordered as desalted DNA oligos.
Create a 10x concentrated primer mix at 20 μM each of these primers in EB buffer.
Add 1 to 4 μl previous PCR product, 2 μl 10x primer mix, 3 to 7 μl water for a total volume of 10 μl, then add 10 μl Kapa HiFi 2x Hotstart PCR master mix and pipette up and down several times.
Use the following cycling parameters: Initial denaturation 98°C 45 sec, denaturation 98°C 15 sec, annealing 60°C 30 sec, extension 72°C 30 sec, final extension 1 min.
Perform a standard Ampure XP/SPRI bead cleanup (e.g. with beads at 1.2x the sample volume) before analyzing the product by microcapillary electrophoresis.
Another graphic illustrating PCR bubbles (source Illumina Inc.). Please also see: https://support.illumina.com/bulletins/2019/10/bubble-products-in-sequencing-libraries–causes–identification-.html
Illumina sequencers using the patterned flowcell technology (NovaSeq X, iSeq, NextSeq 2000, HiSeq 4000 &X Ten) can show an increased rate of barcode switching events. These artifacts are enabled by the exclusion amplification chemistry used on these sequencers; IIlumina calls the artifacts “index hopping”. Please see the index hopping information from Illumina here and in this video from Illumina. We have also have posted additional information here.
Please note that the AVITI sequencers, the MiSeq sequencer and NextSeq 500 chemistries are not sensitive to index hopping. For these sequencers single-indices and combinatorial indexing are fine.
To best avoid barcode switching read mis-assignments on new Illumina sequencers two measures are recommended:
- The use of uniquely dual-indexed (UDI) adapters. Illumina has collected information on the design of such adapters here. This adapter design employs unique barcodes for both index 1 and index 2 for each of the pooled libraries. Thus, 96 samples will use 96 different index 1 adapter sequences and 96 different index 2 adapter sequences.
- The efficient removal of any free primers and adapter-dimers from the libraries
The use of UDI adapters is highly recommended especially for the NovaSeq. Hereby, matching i5 and i7 indices per library must be avoided. UDI adapters can also be used on the MiSeq and the NextSeq, but they do not offer any significant advantages on these sequencers that employ the bridge-amplification chemistry. Further, any traces of free primers, primer-dimers, and adapter dimers should be removed from the sequencing libraries or the pools.
Commercial sources of UDI adapters: TruSeq-style uniquely indexed adapters are available from both Illumina and BiooScientific. Qiagen, NEB, and NuGEN are also supporting their library prep kits with optional UDI adapters. Nextera style indices need to be custom ordered from oligo vendors.
The DNA Technologies Core has 96-plex UDI adapter sets in stock that can be added to sequencing libraries by PCR. These barcode sets are available for both Nextera and TruSeq adapter designs. Please note that for TruSeq style libraries one will need to ligate a shortened and index-less stub-adapter instead of a standard Illumina adapter. The indices are then added after the cleanup of the ligation reaction by PCR.
Sequencing libraries prepared by the DNA Technologies Core:
The DNA Technologies Core uses UDI adapters for all library prep protocols that are compatible with dual indexing (e.g. DNA-Seq, RNA-seq, 3′-Tag-Seg, WGBS-Seq, …). The Core also makes sure to remove any traces of free primers, primer-dimers, and adapter-dimers.
- The fragment lengths should be consistent and best be between 100 and 300 bp (up to 400 bp for the majority of molecules is acceptable). Consistent fragment lengths can best be achieved on a Covaris style closed tube sonicator. We recommend avoiding probe sonicators.
- Please make sure to run the input controls on a Bioanalyzer or agarose gel beforehand, and email us an image of these.
- Sequence one “input control” per cell line/sample type.
- Analyze at least two biological replicates.
- We highly recommend verifying the enrichment of your regions of interest (e.g. promoter regions) vs. the control samples by qPCR, before submitting the samples for sequencing.
- For highest accuracy data we can now generate sequencing libraries with UMI-bearing sequencing adapters. UMIs (Unique Molecular Identifiers) allow the accurate detection and removal of PCR duplicate reads. This approach is especially recommended for low-input samples. The first nine bases of the forward and reverse reads will contain UMI sequences.
The required read number per sample will vary from target to target. For the study of point source transcription factors the ENCODE project recommends analyzing at least 20 million (uniquely mapping) reads (http://genome.cshlp.org/content/22/9/1813.long#boxed-text-2). Depending on the quality of your preps, perhaps 75% of the reads can be expected to be uniquely mapping. ENCODE tends to err on the high side with their recommendations. Thus, about 20 million read pairs per sample should be acceptable, but this is likely the minimum number.
Zhang et al. 2016 have studied the impact of the sequencing run types on ChIP-seq data analysis. Their data indicate that paired-end sequencing data provide significant advantages of single-end sequencing in ChIP-seq.
CUT&RUN sequencing might be a better alternative:
CUT&RUN sequencing (Skene & Henikoff 2017) is a faster protocol that for almost all applications is a more sensitive alternative requiring much lower cell numbers. CUT&RUN is suitable for studying histone modifications, transcription factors, and co-factors. In addition to lower input requirements CUT & RUN experiments also afford reduced read numbers (4 to 8 million read pairs per sample).
References:
Landt et al. 2012: ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Research 22: 1813-1831
Bailey et al. 2013: Practical Guidelines for the Comprehensive Analysis of ChIP-seq Data. PLOS Computational Biology https://doi.org/10.1371/journal.pcbi.1003326
Skene & Henikoff 2017: An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. https://doi.org/10.7554/eLife.21856
Zhang et al. 2016: Systematic evaluation of the impact of ChIP-seq read designs on genome coverage, peak identification, and allele-specific binding detection. BMC Bioinformatics volume 17, Article number: 96
MACS — Model-based Analysis of ChIP-Seq https://taoliu.github.io/MACS/
https://hbctraining.github.io/Intro-to-ChIPseq/lessons/05_peak_calling_macs.html
It is generally recommended to sequence 50 million or more reads/library-molecules per ATAC-seq sample for open chromatin detection and differential analysis (Buenrostro et al. 2015) and 200 million reads for TF footprinting (Yan et al. 2020). Preferred are paired-end sequencing data but single-end data are also usable. Most frequently ATAC-seq libraries are sequenced on the NextSeq with PE 75bp reads.
Paired-end data have slightly higher unique alignment rates, allow for PCR-duplicate removal and provide more complete information about the accessible sequences (due to the longer sequence information). Paired-end data allow for example the assignment of reads to categories such as nucleosome-free, mono-nucleosomal, and di-nucleosomal origins (Buenrostro et al. 2015) .
Controls are typically not run for ATAC-seq studies, but typically two biological replicates are required.
We generate libraries for sequencing with Illumina and PacBio instruments. Illumina libraries include genomic DNA, RNA-seq, ChIP-seq, micro RNA, small RNA, Methyl-seq, RRBS, and reduced representation libraries suitable for GBS analyses, Tag-Seq, and Tn-Seq. Please inquire with us if your protocol is not currently listed. Please see the Library Prep page and the Sample Requirements page for details. We carry out library QC via Bioanalyzer, library size-selections, pooling of sequencing libraries, and real-time quantitative PCR to accurately measure the concentration of sequence-able library molecules to achieve optimal sequencing output.
All sequencing data will be available for secure download via our SLIMS server.
Illumina sequencing data will be delivered as compressed FASTQ files. By default the data will be de-multiplexed (e.g. split according to sample). Each SLIMS directory will further contain a file with the de-multiplexing metrics and a file listing md5 checksums for all the FASTQ files. The checksums can be used to verify that the data integrity has been preserved your download/data transfer.
You will receive all the full-length reads passing the Illumina quality filter. Please contact us if you would like to receive the data quality-trimmed, adapter-trimmed, or adapter-filtered.
For PacBio sequencing you will receive all the data generated by the PacBio SMRT-Portal pipeline (raw data and primary analysis data) including FASTA and FASTQ files as well as the “bax.h5” and “.xml” files required to re-run the analyses. By default we are running also subread filtering as the secondary analysis (except for large gnome projects where this is not required) or other appropriate secondary analysis pipelines for bacterial genome assembly, amplicon analysis, or Iso-seq analyses. You will receive the complete secondary analysis data sets.
Please note: We do NOT archive sequencing data generated for you. Any sequencing data should be downloaded and verified as soon as possible.
The sequencing data will be available for download for three months after they are generated.
Work in the Core is performed on a “first come, first served” basis. HiSeq sequencing data will typically be delivered within about two to five weeks after library submission, while MiSeq has a 5-8 day turnaround time. The turnaround time can fluctuate and is dependent on the number of customer samples in the queue and the read types and lengths requested. Similarly library prep projects will be grouped according to the requested library prep protocols to allow for efficient processing.
Unfortunately not. The samples are put into library prep and sequencing queues after we have received both, the submission form and the sample. Submission forms have to be submitted in hard-copy (accompanying the samples) and electronically (please email the spreadsheet forms to us).
DNA samples and sequencing libraries will be stored at -20 degrees. RNA samples will be stored at -70 degrees C. However samples and libraries will only be stored for one year after sample submission. Please notify us two days in advance if you plan to pick up your samples. Please arrange the sample pick-up within two months of the data delivery. Please provide a FedEx account number and a shipping address if your samples should be shipped back. If possible submit aliquots of your samples to our cores for analysis and store a backup aliquot in your lab.
Please note that custom sequencing primers are not supported by Illumina — the company will not replace sequencing kits on run failures. Nevertheless, custom sequencing primers can enable some unique assays. It is certainly worth exploring if assays can’t be converted to use standard Illumina sequencing primers since standard primers are more or less guaranteed to work. Please also see the amplicon sequencing FAQ and note that the client carries the responsibility for any failures due to bad custom primer oligos or poorly designed custom primers.
On MiSeq sequencers custom sequencing primers can be used for three of the reads (forward, reverse, & 1st index reads)
On the NextSeq 550, custom sequencing primers can be used for all four reads (forward, reverse, & 1st and 2nd index reads)
Illumina does not support custom primers on the NovaSeqs at all.
For Illumina Sequencers: Please provide an aliquot for each custom primer (10 ul at 100 uM in EB buffer; low-bind tubes) with each library submission. Custom sequencing primers should be ordered HPLC purified to remove any incomplete oligos.
For the Element Bio AVITI sequencer: Please provide an aliquot for each custom primer (30 ul at 100 uM in low-TE buffer for index 1, index 2 and read 2 sequencing primers; 40 ul at 100 uM in low-TE buffer for read 1 sequencing primers ; low-bind tubes) with each library submission. Custom sequencing primers should be ordered HPLC purified to remove any incomplete oligos.
When designing custom sequencing primers, the melting temperature needs to be considered (among other criteria). It is suggested to calculate it with the IDT oligo analyzer for the default buffer conditions (50 mM Na+). Please see the recommended melting temperatures in the image below. While at the oligo analyzer site also check the designs for secondary structures, potential hairpin and self-dimers. In case the melting temperatures of your design are too low, the Tm can potentially be increased by inserting interspersed LNA bases into custom LNA oligos available from Qiagen. Avoid clustering of LNA bases and do not substitute the two last 3′ bases. LNA oligos are significantly more expensive than conventional oligos.
For more info please see this guide from Illumina: miseq-system-custom-primers-guide-15041638-01 and also the index read guide: indexed-sequencing-overview-guide-15057455-04-Illumina-pages1to8
Please note that the 2nd-index read is primed from a flowcell-bound oligo for the Miseq and most other Illumina sequencers. Thus, it cannot be customized. The sequencing of the second index begins with exactly seven dark cycles for which no sequence is recorded. Thus, the first base of the 2nd index sequence has to be the 30th nucleotide from the P5 end of the library molecule for most applications. Please note this FAQ can’t be a comprehensive guide to custom sequencing primer design and usage.
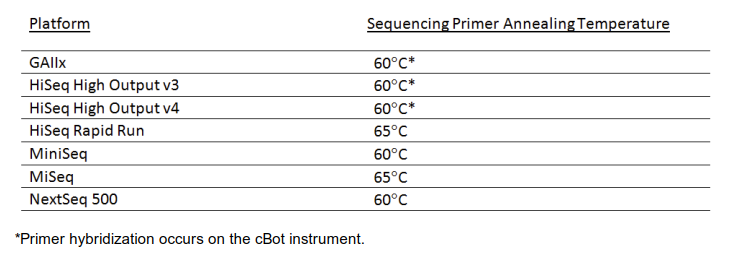
Small RNA-seq / miRNA-seq on the AVITI with libraries employing TruSeq Small RNA sequencing adapters:
Truseq Small RNA libraries will require custom sequencing primers on the AVITI. In most cases there will be no need for a reverse read, thus only TruSeq Small RNA Read 1 and i7/Index 1 custom sequencing primers will be required.
10x Specific recommendations: If using 10X Index Primer Set TS to complete their libraries (i.e. Single Cell Gene Expression Flex or Visium FFPE), they will need to spike-in a TruSeq Small RNA sequencing primer.
| Primer Name | Sequence (5′ -> 3′) | Tm (C) | Length (nt) |
| TruSeq Small RNA Read 1 | ATCTACACGTTCAGAGTTCTACAGTCCGACGATC | 65 | 34 |
| TruSeq Small RNA Read 2 | GTGACTGGAGTTCCTTGGCACCCGAGAATTCCA | 68 | 33 |
| i7/Index 1* | TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC | 68 | 33 |
| i5/Index 2 | Use Adept Index 2 Primer | — | — |
Common buffers for DNA samples, RNA samples, and primers are:
EB-Buffer: 10mM TRIS (pH= 8.0-8.4) – e.g. Qiagen EB Buffer
EBT-Buffer: 10mM TRIS, 0,1%Tween20 (pH=8.0-8.4)
TE-Buffer: 10mM TRIS, 1 mM EDTA (pH=8.0-8.4)
Low-TE buffer or TLE-Buffer: 10mM TRIS, 0.1 mM EDTA (pH=8.0-8.4)
All should be free of DNAses and RNAses.
The DNA Technologies and Expression Analysis Cores do not offer Sanger DNA Sequencing, but these services are available on campus. See the UC DNA Sequencing Facility or the CAES Genomics Facility for more information. There are multiple other service facilities that may be of interest, including the Veterinary Genetics Laboratory, CCM Mouse Biology Program, and Real-time PCR Research and Diagnostics Core Facility. If we missed out any UC Davis facilities that you would like us to list here, please contact us.
06 Sequencing Data (13)
By default you will receive gzip compressed FASTQ data, as individual files for each sample (demultiplexed). The demultiplexing is included in the service if you provide us the barcodes sequences on the submission form.
The files will be available for download from our secure SLIMS server.
You will receive only the reads from clusters passing the Illumina quality filter, also called Illumina chastity filter — please see detailed info below. You will find older recommendations on the internet to also analyze reads from clusters that do not pass the chastity filter. These recommendations are outdated. The filtering is very reliable since several years and it is more or less impossible to find any usable data in the reads that have been filtered out.
Otherwise, the data will be complete. By default we do not trim the sequencing data. We would recommend any quality or adapter trimming to be carried out with third-party tools since they provide better results than the Illumina tools and since there are multiple processing options. SRA submissions also require full-length data.
Please note that the sequence data can contain traces of the Illumina PhiX internal standard. For applications like genome assemblies, these PhiX reads should be removed. BBduk is a free software to achieve this. Please see the Kmer-filtering paragraph in the BBduk help. Please also see the preprocessing section in this presentation: https://ucdavis-bioinformatics-training.github.io/2017-June-RNA-Seq-Workshop/tuesday/Preprocessing.pdf
The Illumina Chastity Filter:
The Illumina chastity filter is applied only to the first 25 bases of the forward read data per cluster. The fluorescence intensity ratios are calculated; specifically the chastity value is defined as the ratio of the brightest base intensity divided by the sum of the brightest and second brightest base intensities. Clusters pass the filter if no more than 1 base call has a chastity value below 0.6 in the first 25 cycles.
Please see this FAQ:
When should I trim my Illumina reads and how should I do it?
Should I trim adapters from my Illumina reads?
This depends on the objective of your experiments. For counting applications such as differential gene expression (DGE), RNA-seq analysis, ChIP-seq, or ATAC-seq, read trimming is generally not required anymore when using modern aligners. For such studies local aligners or pseudo-aligners should be used. Modern “local aligners” like STAR, BWA-MEM, HISAT2, will “soft-clip” non-matching sequences. Pseudo-aligners like Kallisto or Salmon will also not have any problem with reads containing adapter sequences.
However, if the data are used for variant analyses, genome annotation or genome or transcriptome assembly purposes, we recommend read trimming, including both adapter and quality trimming.
DNA and RNA sequencing:
Truseq forward read: AGATCGGAAGAGCACACGTCTGAACTCCAGTCA
Truseq reverse read: AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT
DNA sequencing:
Nextera: CTGTCTCTTATACACATCT
For small RNA/miRNA sequencing data please use this sequence, but also see this FAQ: How should the miRNA/smallRNA data be trimmed?.
TruSeq Small RNA: TGGAATTCTCGGGTGCCAAGG
Should I remove PCR duplicates from my RNA-seq data?
The short and generalized answer to the question “Should I remove PCR duplicates from my RNA-seq data?” is in most cases NO. For some scenarios, de-duplification can be helpful, but only when using UMIs. Please see the details below.
The vast majority of RNA-seq data are analyzed without duplicate removal. Duplicate removal is not possible for single-read data (without UMIs). De-duplification is more likely to cause harm to the analysis than to provide benefits even for paired-end data (Parekh et al. 2016; below). This is because the use of simple sequence comparisons or the typical use of alignment coordinates to identify “duplicated reads” will lead to the removal of valid biological duplicates. RNA-seq library preparation involves several processing steps (e.g. fragmentation, random priming, A-tailing, ligation); none of these processes is truly random or unbiased. Thus, the occurrence of “duplicated reads” in between millions of reads can be expected even in paired-end read data. Short transcripts and very highly expressed transcripts will show the majority of such “natural” duplicates. Their removal would distort the data. For example plant RNA-seq data often seem to contain large amounts of duplicated reads. This is in part due to the fact the gene expression in many plant tissues, like leaves, is dominated by a small number of transcripts; much more so than in most animal samples. Another concern is that the fraction of reads identified as “duplicated” is correlated to the number of aligned reads. Thus, one would have to normalize any data set for equal read numbers to avoid introducing additional bias.
Several studies (among them Parekh et al. 2016; below) have shown that retaining PCR- and Illumina clustering duplicates does not cause significant artifacts as long as the library complexity is sufficient. The library complexity is in most cases directly related to the amount of starting material available for the library preparation. Chemical inhibitors present in the sample could also cause low conversion efficiency and thus reduced library complexities.
PCR duplicates are thus mostly a problem for very low input or for extremely deep RNA -sequencing projects. In these cases, UMIs (Unique Molecular Identifiers) should be used to prevent the removal of natural duplicates. UMIs are for example standard in almost all single-cell RNA-seq protocols.
The usage of UMIs is recommended primarily for two scenarios: very low input samples and very deep sequencing of RNA-seq libraries (> 80 million reads per sample). UMIs are also employed for the detection of ultra-low frequency mutations in DNA sequencing (e.g. Duplex-Seq). For other types of projects, UMIs will have a minor effect in reducing PCR amplification induced technical noise.
Our 3′-Tag-RNA-Seq protocol employs UMIs by default. For other RNA-seq applications please request UMIs on the submission form. When using UMIs for conventional RNA-seq, genomic DNA-sequencing, or ChIOP-seq, the first eleven bases of both forward and reverse reads will represent UMI and linker sequences. These are then followed by the biological insert sequences. The UMI sequences are usually trimmed off and the information transferred into the read ID header with software utilities like UMI-Tools.
Please see the discussion here for details:
https://www.biostars.org/p/55648/ and these excellent papers
Parekh et al 2016: The impact of amplification on differential expression analyses by RNA-seq. and
Fu et al. 2018: Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers.
Kennedy et al 2015: Detecting ultralow-frequency mutations by Duplex Sequencing.
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
This blog post “molecularecologist.com/2016/08/the-trouble-with-pcr-duplicates” offers a detailed analysis of the effect of increasing read numbers on the frequency of PCR duplicates as well as the occurrence of false-positive duplicate identifications on another type of Illumina sequencing data (RAD-seq). Please note that the library type studied is different from RNA-seq as are the potential effects of PCR duplicates for this type of analysis. In contrast to RNA-seq, PCR duplicates should be removed for most RAD-seq studies.
FASTQC is primarily designed to QC whole-genome shotgun sequencing data. Importantly, it is significantly limited in its analyses because it only works on single reads instead of read-pairs.
As a consequence FASTQC tends to generate unnerving warnings for multiple Illumina sequencing data types; This often includes unnecessary warnings about sequence duplication levels and overrepresented sequences.
More modern tools like HTStream and FASTP use more sophisticated algorithms for these purposes and also work on paired-end read data. We highly recommend analyzing duplication levels with these newer tools.
Why does FASTQC show unexpectedly high sequence duplication levels (PCR-duplicates)?
The main reason is that FASTQC only considers single-end sequencing data. All DNA-seq and RNA-seq library preparation protocols involve multiple processing steps. Physical, chemical and enzymatic reactions, all have some sequence-specific biases. As a consequence, the read-start sites are not distributed perfectly random across genomes or transcripts and analyzing a single read start site is insufficient to determine duplicates. Thus, the rate of duplicate reads will always be overestimated and be significantly higher than assumed from read coverage data. Exacerbated is this phenomenon for RNA-seq data which are often dominated by the transcripts of a few genes. For example, it is not uncommon that 50 % of all reads align to the ten most highly expressed genes. The extremely high-read coverage for the particular highly expressed transcripts for RNA-seq data can easily lead to FASTQC read duplication levels of 70% or higher.
Much more realistic read duplication levels can be estimated when incorporating two data points, the read start sites for both forward and reverse reads into the analysis as it is done by HTSream and FASTP.
In addition to only analyzing single reads, FASTQC only analyses 50 nt of the first 100,000 reads for each file for the duplication analysis and extrapolates the dedication rates from this limited number of reads. (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/Help/3%20Analysis%20Modules/8%20Duplicate%20Sequences.html)
Please also see these FAQs:
Should I remove PCR duplicates from my RNA-seq data?
and
What are UMIs and why are they used in high-throughput sequencing?
_____
The deviations from the base composition averages at the start of the sequencing data indicate sequence-specific biases of the library preparation chemistry (here Nextera tagmentation data, which show the most obvious bias).
Please note that the UMIs provide optional additional data analysis options; for many applications, the UMI information can be safely ignored. UMIs are especially beneficial for low RNA input situations as well as ultra-deep sequencing.
For push-button type secondary analyses (combining data for up to 2 SMRT-cells e.g. for demultiplexing, CCS, long amplicon, or IsoSeq analysis) we can run these on our own server and will also deliver all the resulting data. Please note that we are not data analysts, and we can not experiment with analysis parameters. Thus, we highly recommend to work with the Bioinformatics Core for comprehensive data analyses. Alternatively it is recommended that you should install the SMRT Tools command line programs which are part of the SMRT-Link package.
The data files generated by the PacBio Sequel are different from the data generated by the PacBio RSII previously. All the data that were before contained in the bas.h5/bax.h5 and accessory formats are now contained in the .bam, .xml, and .pbi files. Please see this page for the detailed format specifications (pacbiofileformats.readthedocs.io/en/5.0/index.html). Specifically the raw data or each SMRT-cell will be in files named .subreads.bam, .subreads.xml, and .subreads.pbi .
The .bam data can be converted to fastq or fasta files with bamtools (please see at the bottom of this page: github.com/PacificBiosciences/PacBioFileFormats/wiki/BAM-recipes) or best with the PacBio tool bam2fastx. “bam2fastx” is part of the free SMRT Tools: pacb.com/support/software-downloads/ .
!). PacBio came to the conclusion that computing the quality scores for the raw data was a waste of time. Apparently the quality scores for the raw data cannot be reliably computed (and consequently these were also ignored for RSII data pipelines). However, usable PacBio quality scores can be generated from consensus data if the project allows (either by CCS or other secondary analysis algorithms: e.g. by alignments all-vs-all). In short the determination of the quality of individual reads is up the downstream analysis pipeline (e.g. the assembler).We deliver sequencing data via two portals: SLIMS for Illumina data, and BioShare for PacBio and Nanopore data. Both portals offer secure access to the data and support several download protocols. The emails that will notify you about new sequencing data on SLIMS will contain download instructions.
The SLIMS download instructions are available here: https://dnatech.genomecenter.ucdavis.edu/archiving-slims-data/
In BioShare the instructions for several download protocols are integrated into the user interface – BioShare will automatically generate the download commands for you.
Since high throughput sequencing data files tend to be big, we recommend downloading the data by running rsync or wget command line tools (built-in on Linux/Mac/Cygwin). Wget can be easily added to Windows10 as part of a full-featured Linux subsystem (including bash, rsync, wget, …) . This Linux can run in parallel with the Windows GUI. Please see instructions here: How to install WSL on Windows 10 .
The easiest way to add command line UNIX/Linux/BASH functionality (including rsync and wget) to other Windows versions is to install the free MobaXterm terminal from here: https://mobaxterm.mobatek.net/
Please note: We do NOT archive sequencing data generated for you. Any sequencing data should be downloaded and verified as soon as possible.
Illumina sequencing data will be available for download for three months after they are generated. Pacbio and Nanopore data will be available for two months.
Be default we will demultiplex all sequencing data from libraries generated by our facility as well as from customer libraries with barcodes sequenced in separate indexing reads (e.g. using Truseq-style adapters). This is in contrast to adapters with old style in-line barcode data. Data for these have to be demultiplexed by the customer.
The de-multiplexing is included in the sequencing recharge rates. There are no additional costs.
Following analysis of each run, users have access to parsed output through the SLIMS server. A SLIMS account will be created for you on your first run, with information about how to access your account distributed via email (you will receive this email before your actual files are available). The main SLIMS page can be reached here.
You can download all your files from SLIMS with your webbrowser (clicking the links) or better with a download manager (e.g for Firefox https://addons.mozilla.org/en-US/firefox/addon/downthemall/ )
However, it is recommended to download with command line tools. E.g. by running “wget” (built-in into Unix/Linux/Mac/Cygwin operating systems, available also for Windows*) with a command like the following:
wget -r -nH -nc -np -R index.html* “http://slims.bioinformatics.ucdavis.edu/Data/Your_RANDOM_STRING/” &
This command will download all files into your current working directory.
You can also archive all your data with RSYNC by following instructions at
http://wiki.bioinformatics.ucdavis.edu/index.php/Archiving_solexa_data .
*The easiest way to get rsync and wget tools on Windows is by installing the free MobaXterm terminal.
Please note: We do NOT archive sequencing data generated for you. Any sequencing data should be downloaded and verified as soon as possible.
Illumina sequencing data will be available for download for three months after they are generated. Pacbio and Nanopore data will be available for two months.
We do NOT archive sequencing data generated for you. Any sequencing data should be downloaded and verified as soon as possible.
The sequencing data will be available to you on our SLIMS server (Illumina data) or Bioshare server (PacBio and Nanopore data). Illumina sequencing data will be available for download for three months after they are generated. Pacbio and Nanopore data will be available for two months.
You will receive emails from SLIMS or Bioshare notifying you as soon as the data are available for download.
It is highly recommended to download and verify your data (especially the demultiplexing results) at your earliest convenience after receiving the email notification.
We are using the PerkinElmer NEXTflex™ Small RNA-Seq kit for the generation of micro RNA and small RNA-seq libraries because it significantly reduces sequence-specific biases in the library preparation. For this purpose the adapters oligonucleotides contain 4 randomized bases at the ligation junctions. These randomized bases should be removed by trimming before mapping the sequence reads. BiooScientific recommends this procedure:
Data Analysis for micro RNA data generated with the PerkinElmer kits:
The 3′ and 5′ adapters included in this kit both contain 4 random bases that will appear immediately 5′ and 3′ to the insert in sequencing data. The presence of these random bases should be considered when choosing an alignment strategy. When using “end-to-end” alignment, we recommend processing data in the following manner: 1. Clip the 3′ adapter sequence (TGGAATTCTCGGGTGCCAAGG ; this sequence fragment is sufficient!). 2. Trim the first and last 4 bases from the adapter-clipped reads. 3. Perform alignments as normal. Alternatively, alignment may be performed in “local” mode.
Please find the full adapter sequences for your reference here: Bioo-Scientific-Small-RNA-Barcode-Indices-v1-1-15 adapters barcodes small RNA micro-RNA miRNA
Strand-Specific RNA-Seq Libraries
RNA-Seq (conventional) after Poly-A enrichment or ribodepletion:
By default we generate strand-specific RNA-seq libraries. Strand-specific (also known as stranded or directional) RNA-seq libraries substantially enhance the value of an RNA-seq experiment. They add information on the originating strand and thus can precisely delineate the boundaries of transcripts in regions with genes on opposite strands.
There are several ways to accomplish strand-specificity. We incorporate dUTP during the second-strand synthesis of the cDNA. The dUTP containing strand will not be amplified by the proofreading polymerase used for library amplification, thus preserving the strand information for RNA-seq.
For single-end sequencing, the resulting data will represent the “anti-sense strand”. When using paired-end sequencing, the forward read of the resulting sequencing data represents the “anti-sense strand” and the reverse read the “sense strand” of the genes (for Trinity transcriptome assemblies the “–RF” orientation flag should be used). Illumina paired-end reads are always inward oriented (with the exception of “jumping” or “mate-pair” libraries).
Tag-Seq:
Tag-seq data are strand-specific and have a “sense-strand” orientation.
small-RNA-Seq/miRNA-seq:
Small RNA-seq data are strand-specific. The forward read of the sequencing data (read 1) is oriented as the reverse complement of the original RNA molecule. For libraries generated with the Revvity (PerkinElmer) Nextflex small RNA kits, the RNA sequence will be both preceded and followed by four bases with a random sequence.